Graft-Versus-Host Disease
21
AT-A-GLANCE
■ Acute graft-versus-host disease (GVHD) is a serious and potentially life-threatening sequelae of allogeneic hematopoietic stem cell transplantation. Skin manifestations range from a mild, asymptomatic morbilliform eruption to full-thickness skin loss resembling toxic epidermal necrolysis. Hepatic involvement is characterized by elevated total bilirubin. GI disease manifests as abdominal pain, nausea/vomiting, and secretory diarrhea.
■ The most important risk factor for chronic GVHD is a history of acute GVHD. Other important factors include human leukocyte antigen incompatibility, older age, female donor/male recipient, and peripheral blood stem cell source (vs bone marrow).
■ Chronic GVHD of the skin may resemble lichen planus, lichen sclerosus, morphea, systemic sclerosis, or eosinophilic fasciitis. The presentation can be remarkably variable, however, and may resemble folliculitis, keratosis pilaris, or psoriasis. Both epidermal and sclerotic skin manifestations may present at sites of trauma.
■ Patients with chronic GVHD may manifest other autoimmune skin diseases, such as vitiligo and alopecia areata.
■ The pathogenesis of chronic GVHD is poorly understood and nearly every organ system is at risk. The skin, oral mucosa, eyes, GI tract, and lungs are most frequently involved. In many cases, organ system disease resembles known autoimmune conditions.
■ Topical steroids and topical calcineurin inhibitors are used to treat mild, skin-limited chronic GVHD, and systemic steroids are first line in the treatment of moderate to severe chronic GVHD.
■ Optimal dermatologic management of chronic GVHD of the skin requires an understanding of other organ involvement, infection status, and cancer relapse risk. Close communication with the transplantation physician and a “team approach” to multispecialty management is needed.
EPIDEMIOLOGY
Approximately 50,000 hematopoietic stem cell transplantation (HCT) procedures are performed worldwide each year for an expanding array of hematologic malignancies and marrow failure syndromes, metabolic disorders, and immunodeficiencies. HCT may use autologous, syngeneic, or allogeneic donor
hematopoietic stem cells (HCs). During autologous transplantation, the patient’s own HCs are returned to the patient following preparative chemotherapy with or without radiation. Allogeneic HCT (allo-HCT) is the transfer of HCs from a related (nonidentical) or unrelated donor to a recipient. Syngeneic transplantation is the transfer of HCs between identical twins. Graftversus-host disease (GVHD) is the primary cause of non–relapse-related morbidity and mortality in allo- HCT, and also rarely occurs following transplantation of solid organs, transfusion of blood products, and autologous transplantation. Transplantation regimens have advanced rapidly since the first successful allo-HCT was performed in 1968.1 Peripheral blood is now the primary source of donor HCs at many transplantation centers because of decreased risks of relapse and improved survival, although bone marrow transplantation is associated with reduced risk of GVHD.2 Reduced intensity and nonmyeloablative conditioning permitted older patients and others who would not tolerate myeloablative chemotherapy a chance for cure with HCT.3
Umbilical cord blood has gained prominence as a stem cell source in both pediatric and adult HCT given the low risks of GVHD and high engraftment rates.4 Donor leukocyte infusions, the administration of additional donor HCs to the recipient weeks or even months after HCT, may be used to augment graft-versus-malignancy effect, but also may trigger GVHD activity.5
These evolving trends in HCT, in conjunction with other known donor/recipient risk variables (Table 129-1), contribute to a wide range of reported GVHD incidence. The degree of human leukocyte antigen (HLA) mismatch between donor and recipient remains the single most important predictor of GVHD.6 A history of acute GVHD, in turn, confers the greatest risk of developing chronic disease.7 Acute GVHD develops in approximately 40% of fully matched sibling donor HCTs and 80% of mismatched unrelated HCT recipients.8 However, changing peritransplantation regimens, including use of cyclophosphamide and antithymocyte globulin has reduced the rate of acute GVHD in recent years to as low as 10%.9 Risk estimates of chronic GVHD also vary widely from 37% to 47%,2 but data from the Centers for International Bone Marrow Transplant Research demonstrate a gradual increase in chronic GVHD incidence over more than a decade.10 Confounding factors, such as improved posttransplantation survival, may be influencing the apparent trend in increasing chronic GVHD incidence.7
Skin involvement is often the first indicator of acute GVHD (81%), followed by GI (54%) and liver disease (50%).12 Similarly, the majority of patients who develop chronic GVHD manifest skin symptoms at some point in their disease course. Although sclerotic involvement
■Human leukocyte antigen (HLA) incompatibility
■Human leukocyte antigen (HLA) incompatibility
■Patient age (elderly > adult > pediatric)
■Patient age (elderly > adult > pediatric)
■Female donor (especially multiparous)/male recipient
■Female donor (especially multiparous)/male recipient
■Stem cell source (peripheral blood > bone marrow > cord blood)
■Stem cell source (peripheral blood > bone marrow > cord blood)
■T-cell replete graft
■T-cell replete graft
■Unrelated donor
■Unrelated donor
■Donor leukocyte infusion
■Donor leukocyte infusion
■Interruption or rapid tapering of immunosuppression
■Interruption or rapid tapering of immunosuppression
■Total body irradiation in pretransplantation conditioning regimen11
■Total body irradiation in pretransplantation conditioning regimen11
is less common than “lichenoid” GVHD and tends to occur later post-HCT, sclerotic features, particularly deep-seated fascial changes, may have an insidious onset, and “lichenoid” involvement is not a prerequisite to the development of sclerotic features. The incidence of sclerosis in chronic GVHD has been reported to be between 3.6% and 22.6%.10,11,13,14
ETIOLOGY AND PATHOGENESIS
21
minor histocompatibility antigens (eg, HY, HA-3).16 An allogeneic response to these minor antigens is thought to partially explain the development of GVHD in 40% of recipients of HLA-identical grafts. Host autoreactive T cells may also play a role in disease pathogenesis.17 Tissue damage, infection, and pretransplantation conditioning modifies the inflammatory response through proinflammatory cytokine production and antigen-presenting cell activation.18
Antigen-presenting cell activation may be mediated by damage-associated molecular patterns and pathogenassociated molecular patterns released from damaged skin, gut, and vascular tissue.19 Demonstration of lichen planus-like and sclerotic chronic GVHD at sites of injury and viral infection has highlighted a likely role for Type I interferons, which are known to play a critical role in the innate immune response to cellular damage and viral infection.20
Acute GVHD has traditionally been considered T-helper cell (Th) 1–mediated, while Th2 mechanisms were thought to predominate in chronic GVHD. It is now known that Th17 pathways also play a role in the pathogenesis of both disease processes.21 Donor interleukin (IL)-17A, derived from Th17 cells, promotes skin fibrosis. There also is increased IL-17 messenger RNA in skin of chronic GVHD patients, as well as higher levels of Th17-promoting cytokines including IL-6 and IL-21, and transcription factor STAT3.21-23
Following activation of host antigen-presenting cells, T-cell activation and differentiation drives the response in acute GVHD. T-cell activation results in a massive release of proinflammatory cytokines, causing damage to the organs and tissues.24 The final effector phase of acute GVHD is characterized by cell damage via cytotoxic T cells, natural killer cells, and soluble inflammatory mediators, including tumor necrosis factor (TNF), interferon-γ, and IL-1.18
Regulatory T cells are known to play a key role in inhibiting GVHD.25 Acute GVHD may target secondary
ACUTE GVHD CHRONIC GVHD
Time to onseta Usually <100 days after transplantation Usually >100 days after transplantation
Immune pathways T-helper (Th)-1 and Th17 mediated Th2 and Th17 mediated
Cellular response T cells (cytotoxic T cells) play a major role; also natural killer cells Decreased regulatory T cells
Inflammatory mediators Tumor necrosis factor Interferon-γ Interleukin (IL)-1 IL-2
Histologic features
B cells play a major role; also T cells, plasma cells Tissue macrophages Decreased regulatory T cells
B-cell activating factor Platelet-derived growth factor Transforming growth factor-β IL-6 IL-21 B-cell autoantibodies (ANA, anti-dsDNA)
Necrotic keratinocytes with dermal lymphocytic infiltrate Basal vacuolar interface alterations or subepidermal cleft forma r - tions or epidermal–dermal separation (depending on severity) r
Vacuolar interface dermatitis with dyskeratotic keratinocytes Sclerosis of the upper dermis Epidermal atrophy, hyperkeratosis, follicular plugging
Histologic features (cutaneous) Necrotic keratinocytes with dermal lymphocytic infiltrate Basal vacuolar interface alterations or subepidermal cleft formations or epidermal–dermal separation (depending on severity)
(cutaneous)
Vacuolar interface dermatitis with dyskeratotic keratinocytes Sclerosis of the upper dermis Epidermal atrophy, hyperkeratosis, follicular plugging
2321
aClassic presentation; however, features and timing may overlap. (See revised classification in Fig. 129-1.) ANA, anti-nuclear antibody; dsDNA, double-stranded DNA; GVHD, graft-versus-host disease.
21
lymphoid organs, which may result in loss of regulatory T-cell production.17 Decreased T-regulatory cells are associated with severity of acute GVHD and poor response to GVHD treatment.26 Although many therapies for acute GVHD target T cells, particularly IL-2 or its receptor (CD25), these approaches may have the unintended consequence of adversely impacting the CD4+CD25+ regulatory T-cell population.27
The pathophysiology of chronic GVHD is less-well understood, but may share the same activating and regulatory mechanisms as acute GVHD.19 Chronic GVHD is also associated with decreased numbers of regulatory T cells.28 Natural killer cells and regulatory B cells may also serve roles in preventing GVHD.29,30
Chronic GVHD is thought to be at least partly mediated through Th2 interaction with B cells and autoantibody induction. B cells may act as antigen-presenting cells, priming T cells to respond to HLA antigens in cases of partial matching, or minor histocompatibility antigens, and high-titers of antibodies directed against minor histocompatibility antigens are associated with chronic GVHD, particularly histocompatibility Y (HY) antigen.31 Similarly, soluble levels of B-cell activating factor (BAFF), a cytokine which inhibits apoptosis of B cells and promotes differentiation into plasma cells, correlate with chronic GVHD activity.32 B-cell autoantibodies have been shown to associate with Th17 cells in skin, and perpetuate GVHD in mice.33
The role of B-cell function in chronic GVHD is supported by reports of success of use of the anti-CD20 antibody rituximab in chronic GVHD.34 Patients who respond well to rituximab have higher circulating levels of activated B cells.35 Administration of rituximab in preparative regimens also has been associated with lower incidence and severity of acute GVHD.36 Autoantibody formation (eg, antinuclear antibody, anti– double-stranded DNA antibody) is a frequent finding in chronic GVHD, although the antibodies lack the specificity of typical autoimmune disease.37
The mechanisms responsible for chronic GVHDinduced fibrosis in the skin and elsewhere (eg, bronchiolitis obliterans) remains uncertain. Platelet-derived growth factor (PDGF) may activate transforming growth factor-β, a potent profibrotic cytokine capable of stimulating collagen production, abrogating metalloproteinase activity, and sensitizing fibroblasts to a constitutive-activated state via autocrine signaling.38-40 Stimulatory antibodies targeting the PDGF receptor have been proposed as the etiology of fibrosis in GVHD and systemic sclerosis.39 However, the significance of these antibodies remains unclear and other studies have failed to correlate PDGFR antibody with severity of chronic GVHD.41 Kinase inhibition of intracellular PDGF signaling is a targeted therapeutic approach in sclerotic chronic GVHD. Macrophages are known to play a role in the tissuerepair response contributing to fibrosis,42 and have been shown to accumulate in fibrotic lesions in chronic GVHD.43 Macrophages in GVHD tissue are of donor origin, and may be efficient in generating antibodies that may contribute to GVHD pathogenesis, including transforming growth factor-β.19 Macrophages may also represent potential therapeutic targets.
2322
In recent years, genome-wide association studies have begun to define individual genetic risk factors for GVHD.44 Studies have revealed that single-nucleotide polymorphisms in NOD2 (nucleotide-binding oligomerization domain-2), TNF, and IL-10 are associated with GVHD. Single-nucleotide polymorphisms in BANK1, CD247, and HLA-DPA-1 are associated with development of sclerotic GVHD.45
CLINICAL FINDINGS
HISTORY
HISTORY
Accurate diagnosis of acute GVHD is challenging, and requires a thorough assessment and clinicopathologic correlation. Because the skin eruption (and histology) may be nonspecific in the context of several potential differential diagnoses, a careful history is invaluable. Key factors include assessment of the primary disease for which the patient underwent an organ transplant procedure, stem cell source (eg, peripheral blood, bone marrow, umbilical cord blood), type of transplant (eg, allogeneic, syngeneic, autologous), degree of HLA match, use of a related versus unrelated donor, and T-cell depletion of the graft. Additional factors include agents used for GVHD prophylaxis and the pretransplantation conditioning regimen. The use of particular chemotherapeutic agents such as cytarabine may render the differential diagnosis of toxic erythema of chemotherapy more likely.46 Reduced intensity conditioning and nonmyeloablative conditioning may be associated with increased risk of sclerotic chronic GVHD,11 and may delay the onset of acute GVHD symptoms beyond the 100-day period.47 The timing of engraftment, new medication exposures, virus status of both the donor and recipient (eg, cytomegalovirus), and evidence of other organ involvement (eg, elevated total bilirubin, diarrhea) provide additional data for clinicopathologic correlation. Bilirubin elevation and diarrhea, however, are nonspecific, and may be related to concomitant viral infection, drug toxicity and bile duct obstruction. Features of acute GVHD following recent blood transfusion should raise concern for transfusionassociated GVHD (TA-GVHD). TA-GVHD is an oftenfatal sequelae of administration of cellular blood products to immunocompromised HCT recipients. As a result, all blood products given to HCT recipients must be irradiated. TA-GVHD may also occur following transfusion of unirradiated blood products to children with congenital immunodeficiency. TA-GVHD may also occur in the immunocompetent setting following transfusion of an unirradiated blood product that contains donor lymphocytes that are homozygous for the HLA haplotype of the recipient, typically when transfusion occurs from a relative or genetically similar person. For example, in Japan where the genetic background of the population is more homogenous, the estimated risk of randomly receiving blood from a homozygous donor is 1 in 874.48 In this form of
TA-GVHD, the donor lymphocytes in the blood product are not recognized as foreign, leading to a GVHD reaction similar to classic acute GVHD. Beginning 10 days after transfusion, fever and skin rash (histologically consistent with GVHD) develops, followed by liver dysfunction and diarrhea. Death from pancytopenia usually occurs within several weeks.49
As with acute disease, a new diagnosis of chronic GVHD is best made based by history, cutaneous examination, and histology. A previous history of acute GVHD is the single greatest risk factor for chronic disease. When there is no cessation of acute GVHD prior to development of chronic GVHD, it is considered progressive, and carries the poorest prognosis. In contrast, quiescent disease is the onset of chronic disease after previous resolution of acute GVHD symptoms. Chronic GVHD is considered de novo when there is no prior history of acute GVHD. Traditionally, acute GVHD occurs within 100 days posttransplantation, while symptoms after 100 days are considered chronic GVHD. However, because acute symptoms may develop after 100 days posttransplantation and chronic symptoms may develop before then, the revised classification of acute and chronic GVHD symptoms includes additional subtypes of GVHD with overlapping features or timing of acute and chronic symptoms (Fig. 129-1).50 Recent tapering of immunosuppressant medication or donor leukocyte infusion given to augment the graftversus-malignancy response are potential triggers of skin activity. Donor leukocyte infusions, in particular, may present with an acute GVHD skin eruption consistent with acute GVHD rather than the eruption of chronic disease. Cutaneous or systemic infection may also induce a flare of skin GVHD, as will drug reactions, which can result in a diagnostic challenge given the clinical and histologic similarities between viral exanthem, drug eruption, and GVHD.51 Intense ultraviolet (UV) exposure may also trigger skin GVHD.
21
Important clues to sclerotic and fascial disease includes a new-onset edema of an extremity, muscle cramping, decreased flexibility, and complaints of skin tightness, particularly at the waistband and brassiere-line,52 and at sites of prior injury or trauma, such as intravenous access sites or radiation fields.53
Although GVHD in other organ systems may not necessarily flare in synchrony with skin involvement, the presence of other organ system involvement is helpful when the cutaneous features are nondiagnostic. Therefore, a thorough review of systems should be conducted to assess for organ involvement. Common GVHD symptoms include oral and ocular sicca and oral pain, particularly with spicy foods. The presence of genital discomfort or pain also suggests GVHD. Dysphagia may indicate the presence of esophageal strictures or webbing. Bronchiolitis obliterans manifests as dry cough, wheezing, and dyspnea, but requires pulmonary function tests and CT scans to rule out infection and other etiologies. Also common, but less specific, are symptoms of fatigue, poor appetite, and weakness.54,55
Finally, it is important to remember that despite the phenotypic variability in chronic GVHD of the skin, not every skin manifestation in a patient after HCT is caused by GVHD, so a careful dermatologic history to detect other possible diagnoses is prudent.
CUTANEOUS PRESENTATION
CUTANEOUS
PRESENTATION
The most common presentation of acute GVHD begins with erythematous-dusky macules and papules of the volar and plantar surfaces and ears that may rapidly become a diffuse morbilliform exanthem (Fig. 129-2A and B; Table 129-3). Very early
Revised classification of acute and chronic GVHD
GVHD manifestation
cGVHD diagnostic feature or distinctive feature (if bx proven) aGVHD feature
≤100 days post-HCT >100 days post-SCT No features of aGVHD Additional aGVHD feature
Classic aGVHD
Delayed aGVHD
Overlap syndrome
Classic cGVHD Recurrent aGVHD Persistent aGVHD
2323
21
A
C
B
D
E
involvement may manifest as erythema limited to hair follicles (Fig. 129-2C). A predisposition for acral and perifollicular sites may help distinguish newonset acute GVHD from other morbilliform eruptions, although these features are not always present.
2324
Pruritus is variable and is also not useful to distinguish acute GVHD from other causes. Erythroderma and, in severe cases, spontaneous bullae (Fig. 129-2D) with skin sloughing resembling toxic epidermal necrolysis may develop (Fig. 129-2E). Widespread erythrodermic
Skin
■Erythema of palms, soles, ears
■Perifollicular erythema
■Generalized exanthem
■Bullae/necrolysis
Gastrointestinal
■Abdominal pain
■Anorexia
■Ileus
■Mucositis
■Vomiting
■Secretory diarrhea
Liver
Liver
■Endothelialitis
■Endothelialitis
■Pericholangitis
■Pericholangitis
■Cholestatic hyperbilirubinemia
■Cholestatic hyperbilirubinemia
21
involvement, particularly skin sloughing, portends a very poor prognosis. Acute GVHD is staged based on percent body surface area involvement with concordant histologic findings, as well as degree of bilirubin elevation and diarrhea (Table 129-4).56 In contrast to chronic disease, postinflammatory pigmentary changes following acute GVHD are uncommon. Clinical features of chronic skin GVHD are classified as diagnostic or distinctive. Diagnostic criteria establish the presence of chronic GVHD without the need for further testing or evidence of other organ involvement, while distinctive criteria warrant skin biopsy or establishment of additional organ involvement (Table 129-5). Diagnostic cutaneous features of GVHD include poikiloderma, lichen planus–like lesions, and sclerotic skin changes.50 There is a growing appreciation of the tremendous variability in the clinical presentation of chronic skin GVHD. Previously, especially in the organ transplant community, skin findings in chronic GVHD were dichotomized as “lichenoid” or
STAGE SKIN HISTOLOGY DIARRHEA (500 mL/day) BILIRUBIN (mg/dL)
0 – – ≤500 ≤2
1 <25% body surface area (BSA) Focal or diffuse vacuolar interface >500 or persistent anorexia, nausea, vomiting 2 to 2.9
2 25% to 50% BSA Dyskeratotic keratinocytes ≥1000 3 to 5.9
3 >50% BSA Subepidermal clefting ≥1500 6 to 14.9
4 Erythroderma with bullae or
Loss of epidermis ≥2000 or severe abdominal pain ± ileus ≥15
4 Erythroderma with bullae or desquamation Loss of epidermis ≥2000 or severe abdominal pain ± ileus ≥15
desquamation
DIAGNOSTIC DISTINCTIVE OTHER
Skin
■Poikiloderma
COMMON (SEEN IN BOTH ACUTE AND CHRONIC GVHD)
■Depigmentation (including vitiligo)
■Lichen sclerosus–like lesions
■Papulosquamous lesions
■Lichen planus–like eruption
■Morphea-like lesions
■Sclerotic features
Nails –
■Dystrophy
■Sweat impairment
■Erythema
■Ichthyosis
■Maculopapular rash
■Keratosis pilaris
■Pruritus
■Hypopigmentation
■Hyperpigmentation
– –
■Longitudinal ridging, splitting, or brittle features
■Onycholysis
■Pterygium unguis
■Nail loss (symmetric, most nails)
Hair –
■New-onset scarring or nonscarring scalp alopecia
■Thinning scalp hair, typically patchy
Hair – ■New-onset scarring or nonscarring scalp
alopecia
■Loss of body hair
■Loss of body hair
■Scaling
■Scaling
■Thinning scalp hair,
–
–
typically patchy
■Coarse or dull hair
■Coarse or dull hair
2325
■Premature gray hair
■Premature gray hair
21
“sclerodermoid.” The term lichenoid GVHD had been used to denote any involvement of the skin in which erythema or scaling was present; however, “lichenoid” is a histologic pattern, not a clinical one, and, therefore, usage of it is best reserved to pathologic description. Furthermore, although chronic GVHD may resemble lichen planus (Fig. 129-3), other epidermal patterns are frequently observed, such as ichthyosis, poikiloderma (Fig. 129-4), and skin lesions resembling lupus erythematosus, keratosis pilaris, or psoriasis.57 A linear form of chronic GVHD resembling lichen planus has been described, which may present following Blaschko lines (Fig. 129-5). Postinflammatory hyperpigmentation is common following the resolution of epidermal involvement, particularly in darkly pigmented individuals, and may persist for many months after the skin disease becomes quiescent. The fibrosing manifestations of chronic GVHD are also remarkably variable, and the term sclerodermoid is an inadequate descriptor of the varied tissue abnormalities in the dermis, subcutaneous tissue, and fascia. As in systemic sclerosis, an edematous phase may herald the onset of skin fibrosis, but in contrast to systemic sclerosis, the face, fingers, and toes are usually spared,
2326
and the typical acral-to-proximal progression characteristic of systemic sclerosis is not commonly seen in chronic GVHD. Certainly, many cases do involve the hands and fingers, but occur more proximal to the fingertips (Fig. 129-6).58 Early superficial fibrotic involvement resembles lichen sclerosus. In contrast to traditional lichen sclerosus, which is much less common on the skin than the genital region, lichen sclerosis in GVHD patients often manifests on the skin as porcelain-white atrophic plaques on the upper back (Fig. 129-7A). A common pattern of GVHDassociated fibrosis involves patchy sclerotic plaques with hypopigmentation and hyperpigmentation mimicking morphea (Fig. 129-7B). Sclerosis of this type
A
C
E
21
B
D
F
2327
21
may exhibit an isomorphic response, localizing to the sites of minor skin trauma, particularly the waistband area,52 or may develop at sites of previous scar formation (Fig. 129-7C) or radiation. It also has been associated with an isotopic response, developing at sites of prior zoster infection (Fig. 129-7D).53 Diffuse dermal involvement may result in a “pipestem” appearance of the lower extremities with marked reduction in limb volume and overlying shiny hidebound skin with loss of hair resembling scleroderma (Fig. 129-7E). Deeper involvement of the subcutaneous fat results in irregular hyperpigmented sclerotic plaques with intervening areas of edematous skin closely resembling deep morphea/morphea profunda.59 Bullae may develop at sites of fibrosis, particularly on the lower legs, as a result of dermal edema, as has been described in bullous morphea profunda.60 Patchy hyperpigmentation (“leopard spots”) may be visible prior to the diagnosis of dermal sclerotic involvement.61 Dermal sclerosis results in decreased ability to pinch the skin. Primary involvement of the subcutaneous fat and fascia results in a diffuse firm, rippled pattern to the skin resembling eosinophilic fasciitis (see Fig. 129-7F).62 Features of overlying epidermal GVHD involvement and pigmentary changes may be absent. Fascial involvement is often most visible on the medial arms and thighs and be accentuated by abduction and supination of the arm. Prominent “grooving” demarcating fascial bundles and along the path of superficial vessels may be observed. Careful palpation of the skin is helpful in detecting deep-seated irregularities in skin texture and differentiation from cellulite. Examination of patients while they are lying down on the examination table may help to reveal rippling or pseudocellulite of the abdomen.54 Dermal fibrosis or fascial involvement without overlying dermal thickening may lead to progressive loss of joint range of motion and contracture formation. Unlike in systemic sclerosis, chronic GVHD tends to affect large joints, including the shoulders, elbows, wrists, hips, and ankles.58 Range of motion may be assessed through simple maneuvers on physical examination including flexion/extension (arms/shoulders), abduction/ adduction (thighs), and prayer sign (wrists, fingers). It may be helpful to have children sit cross-legged on the examination table to determine range-of-motion abnormalities of the hips.54 Fasciitis may be associated with cramping, polymyositis, and arthritis. Nail involvement in chronic GVHD typically results in longitudinal ridging and thin, easily broken nails. Partial or complete anonychia and dorsal pterygium formation may occur.50 Other findings include onycholysis, periungual erythema, and paronychia.63
Other skin sequelae of chronic GVHD include milia formation, porokeratosis, often on the buttock area,64 angiomatosis,65 nipple hyperkeratosis,66 vitiligo, and alopecia, either diffuse or focal areas of alopecia areata.67 A verrucous acral keratosis form of GVHD has been described, resembling common warts on the fingers, but distinguished by the presence of vacuolar interface and dyskeratotic keratinocytes on histology.68
Multiple manifestations of sclerotic and nonsclerotic skin disease may be present in the same
2328
individual, making accurate quantification of disease activity challenging. A number of different instruments are currently under investigation to measure skin involvement, most commonly the National Institutes of Health (NIH) Consensus organ-specific severity scoring system, in which the skin composite score in separated into body surface area and specific skin features.50 Other tools include the Vienna Skin Score,69
the Hopkins skin sclerosis and fasciitis scores,70 and the Lee symptom scale and skin subscale.55 The modified Rodnan score, which is validated for use in systemic sclerosis,71 has not been validated for use in sclerotic GVHD, and may be of limited value for fascial disease.
RELATED PHYSICAL FINDINGS
RELATED PHYSICAL
FINDINGS
Acute GVHD is primarily a disorder of the skin, GI tract, and liver (see Table 129-3), typically presenting with skin rash, new-onset elevation of total bilirubin, and/or voluminous diarrhea. By contrast, chronic GVHD is remarkably diverse in its breadth of organ system manifestations (Table 129-6). The most frequently affected sites are skin and nails, oral mucosa, eyes, liver, lungs, and marrow (usually thrombocytopenia).7 Esophageal webs/strictures, vaginovulvar disease, myositis, nephrotic syndrome, and pericarditis are less-frequent sequelae of chronic disease. Mucosal disease is second only to skin involvement in frequency in chronic GVHD. Erythema, lichen planus–like changes with Wickham striae, erosions and ulcerations, and mucoceles are the most common manifestations. The buccal mucosa is most commonly affected, followed by the lips, tongue, and soft palate. Patients often also experience sicca symptoms, and pain from erosions may limit oral intake.54 Dryness and violaceous erythema of the lips are common. Genital involvement significantly impairs sexual function and quality of life and may be overlooked if a specific examination and directed questions regarding genital symptoms are not undertaken. Involvement of the penis may induce phimosis. Vulvovaginal involvement presents as erythema, erosions/fissures, vestibulitis, vaginal stenosis, labial resorption, or complete agglutination of the introitus leading to hematocolpos (Fig. 129-8).72
The 2014 chronic GVHD NIH Consensus Development Project provided precise terminology for organspecific diagnostic manifestations of chronic GVHD in the setting of HCT (see Table 129-6).50
HISTOPATHOLOGY
HISTOPATHOLOGY
21
Mucosal Involvement Oral mucosa
Mucosal Involvement Oral mucosa
■Erythema
■Erythema
■Gingivitis
■Gingivitis
■Lichen planus-likea
■Lichen planus-likea
■Mucocele
■Mucocele
■Mucosal atrophy
■Mucosal atrophy
■Mucositis
■Mucositis
■Pain
■Pain
■Pseudomembrane
■Pseudomembrane
■Ulcer
■Ulcer
■Xerostomia Genital mucosa
■Xerostomia Genital mucosa
■Lichen planus–likea
■Lichen planus–likea
■Lichen sclerosus–likea
■Lichen sclerosus–likea
■Phimosis or urethral meatus scarring or stenosisa
■Phimosis or urethral meatus scarring or stenosisa
■Vulvar erosions/fissures/ulcers
■Vulvar erosions/fissures/ulcers
■Vaginal scarring or clitoral/labial agglutinationa
■Vaginal scarring or clitoral/labial agglutinationa
Muscle, Fascia, Joint Involvement
Muscle, Fascia, Joint Involvement
■Arthralgia or arthritis
■Arthralgia or arthritis
■Edema
■Edema
■Fasciitisa
■Fasciitisa
■Joint stiffness or contractures secondary to fasciitis or sclerosisa
■Joint stiffness or contractures secondary to fasciitis or sclerosisa
■Muscle cramps
■Muscle cramps
■Myasthenia gravis
■Myasthenia gravis
■Myositis or polymyositis Other Organ System Involvement Cardiovascular
■Myositis or polymyositis Other Organ System Involvement Cardiovascular
■Pericardial effusion
■Pericardial effusion
■Cardiac conduction abnormality or cardiomyopathy
■Cardiac conduction abnormality or cardiomyopathy
Ophthalmologic
Ophthalmologic
■Blepharitis
■Blepharitis
■Cicatricial conjunctivitis
■Cicatricial conjunctivitis
■Confluent punctuate keratopathy
■Confluent punctuate keratopathy
■Dry, gritty, or painful sensation in eyes
■Dry, gritty, or painful sensation in eyes
■Periorbital hyperpigmentation
■Periorbital hyperpigmentation
■Photophobia GI
■Photophobia GI
■Anorexia, nausea, vomiting, diarrhea, weight loss, failure to thrive
■Anorexia, nausea, vomiting, diarrhea, weight loss, failure to thrive
■Esophageal weba
■Esophageal weba
■Esophageal upper third stricture/stenosisa
■Esophageal upper third stricture/stenosisa
■Exocrine pancreatic insufficiency Hematopoietic and immune
■Exocrine pancreatic insufficiency Hematopoietic and immune
■Autoantibodies (autoimmune hemolytic anemia, immune thrombocytopenic purpura)
■Autoantibodies (autoimmune hemolytic anemia, immune
thrombocytopenic purpura)
■Eosinophilia
■Eosinophilia
■Hypogammaglobulinemia/hypergammaglobulinemia
■Hypogammaglobulinemia/hypergammaglobulinemia
■Lymphopenia
■Lymphopenia
■Thrombocytopenia
■Thrombocytopenia
■Raynaud phenomenon Hepatic
■Raynaud phenomenon Hepatic
■Elevated total bilirubin
■Elevated total bilirubin
■Elevated alkaline phosphatase
■Elevated alkaline phosphatase
■Elevated transaminases Neurologic
■Elevated transaminases Neurologic
■Peripheral neuropathy Pulmonary
■Peripheral neuropathy Pulmonary
■Air trapping and bronchiectasis on CT
■Air trapping and bronchiectasis on CT
■Bronchiolitis obliterans diagnosed by biopsya or syndrome
■Bronchiolitis obliterans diagnosed by biopsya or syndrome
■Cryptogenic organizing pneumonia
■Cryptogenic organizing pneumonia
■Pleural effusions
■Pleural effusions
■Restrictive lung disease Renal
■Restrictive lung disease Renal
■Nephrotic syndrome
■Nephrotic syndrome
aDiagnostic features of chronic graft-versus-host disease (GVHD) based on NIH Consensus Criteria. Other signs and symptoms listed are not considered sufficient to establish a diagnosis of chronic GVHD without further testing or evidence of other organ system involvement. The most common GVHD manifestations are shown in bold. Adapted from Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graftversus-host disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant. 2015;21:389-401.e1.
presence of necrotic keratinocytes accompanied by a dermal lymphocytic infiltrate (usually sparse) and basal vacuolar interface alteration (Fig. 129-9). Early GVHD involvement with follicular erythema correlates with involvement limited to the hair follicle. Subepidermal cleft formation (grade III) is indicative of more-severe involvement, whereas complete separation of epidermis from dermis (grade IV) correlates with clinical findings resembling toxic epidermal necrolysis. Grade IV involvement may be impossible to differentiate histologically from drug-induced toxic epidermal necrolysis and requires careful clinical correlation. The presence of eosinophils has been used to argue against a diagnosis of GVHD; however, it cannot be reliably used to distinguish between GVHD and drug hypersensitivity.73 Engraftment syndrome is a poorly understood phenomenon at the time of neutrophil engraftment following autologous HCT or allo-HCT and is characterized by a nonspecific erythematous skin eruption, fever, and
pulmonary edema.74 Histologically, it may not be possible to distinguish engraftment rash from early (grade I) acute GVHD. Epidermal changes in chronic GVHD may be indistinguishable from those of acute disease and are characterized by vacuolar interface dermatitis and epidermal dyskeratotic keratinocytes (Fig. 129-10A). Adnexal structures are often affected. Sclerotic involvement of the upper dermis may resemble lichen sclerosus, with epidermal atrophy, hyperkeratosis, follicular plugging, and a pale, homogenized appearance of the upper dermis collagen (Fig. 129-10B). Sclerotic GVHD is characterized by thickened collagen bundles, increased numbers of fibroblasts, loss of periadnexal fat, and periadnexal inflammation, and may be indistinguishable from morphea and scleroderma. Subcutaneous and fascial involvement accordingly demonstrates changes in the fat septae and fascia, including thickening, edema, and fibrosis.
2329
21
An inflammatory infiltrate, consisting of variable numbers of lymphocytes, histiocytes, and eosinophils may be seen.57,75
Histology of oral mucosal GVHD reflects similar interface changes as those seen in epidermal GVHD, but without associated acanthosis. Lymphocytic
infiltration of the salivary glands resembles changes seen in Sjögren syndrome.75,76
DIAGNOSIS
LABORATORY TESTS
LABORATORY TESTS
Diagnosis of acute GVHD skin involvement is based on clinicopathologic correlation, particularly exclusion of drugs and infectious causes. Total bilirubin levels and quantification of diarrhea volume are used in conjunction with skin disease to stage the disease (see Table 129-3). The presence of a neutrophil count greater than 0.5 × 109/L and a platelet count greater than 20 × 109/L indicates engraftment, and brings engraftment syndrome into the differential diagnosis of acute GVHD.74
In recent years, genome-wide association and proteomic studies have helped identify potential biomarkers in GVHD patients. Candidate markers found to
A B
2330
have diagnostic and prognostic value for acute GVHD include TNF receptor-1, IL-2 receptor-α, IL-8, hepatocyte growth factor, and regenerating islet-derived 3α. Elevated levels of these markers have been associated with increased likelihood of nonresponse, as well as poorer survival.77 Additional markers whose increased expressed may be associated with acute GVHD include BAFF, IL-33, CXCL-10, and CXCL-11.78
In chronic GVHD, circulating B-cell subsets and decreased T-regulatory cells are associated with disease.28,37,69,79 BAFF, CXCL-9, elafin, aminopeptidase N, soluble-type IL-2 receptor-α, IL-4, IL-6, and TNF also are associated with diagnostic usefulness in chronic GVHD.80-82 TNF has shown both diagnostic and prognostic value in chronic disease.83 Although autoimmune markers may be seen in the majority of patients after allo-HCT, their presence is generally not specific for the development of chronic GVHD manifestations. In an NIH cohort, autoantibodies were identified in 62% of chronic GVHD patients, and multiple antibodies in 35% of patients. The most frequent antibodies were antinuclear antibody (29%) and rheumatoid factor (13%).37 Other autoantibodies that have been reported in GVHD patients include anti–doublestranded DNA, anti-PDGF, and anti-HY.31,39,84 Identifying specific biomarkers of disease activity is an area of research emphasis in acute and chronic GVHD.82
SPECIAL TESTS (INCLUDING IMAGING STUDIES)
SPECIAL TESTS (INCLUDING
IMAGING STUDIES)
Suspicion of subcutaneous sclerotic and fascial disease and myositis may be confirmed by MRI, particularly in cases in which definitive sclerotic changes are not observed or when a fascial or muscle biopsy is deferred.85
High-frequency ultrasound,86 digital heat mapping, and durometry also have been used to study sclerosis of the skin in GVHD, but are not widely available in clinical practice.
DIFFERENTIAL DIAGNOSIS
The differential diagnosis of acute GVHD is broad, and especially challenging as the clinical and histologic features are often indistinguishable from morbilliform drug eruption and engraftment syndrome, the latter of which follows neutrophil recovery and presents with a morbilliform eruption, fever, and pulmonary edema. It also may be accompanied by elevated total bilirubin and diarrhea, and appears histologically similar to acute GVHD.74 Although engraftment syndrome is thought to occur sooner posttransplantation than GVHD, hyperacute GVHD can present within this window as well. Engraftment syndrome tends to be more steroid-responsive compared to acute GVHD. Toxic erythema of chemotherapy may occur up to 1 week after transplantation, and encompasses multiple
21
Acute Graft-Versus-Host Disease (GVHD)
■Drug eruption
■Eruption of lymphocyte recovery
■Rash of engraftment syndrome
■Transient acantholytic dermatosis
■Toxic epidermal necrolysis (for stage IV disease)
■Toxic erythema of chemotherapy
■Viral exanthem
Chronic GVHD Epidermal involvement
Chronic GVHD Epidermal involvement
■Drug eruption
■Drug eruption
■Demodex folliculitis
■Demodex folliculitis x x
■Eczematous drug eruption
■Eczematous drug eruption
■Lichen planus
■Lichen planus
■Pityriasis lichenoides chronica
■Pityriasis lichenoides chronica
■Pityrosporum folliculitis
■Pityrosporum folliculitis
■Psoriasis Sclerotic involvement
■Psoriasis Sclerotic involvement
■Eosinophilic fasciitis
■Eosinophilic fasciitis
■Lichen sclerosus
■Lichen sclerosus
■Morphea
■Morphea
■Nephrogenic systemic fibrosis
■Nephrogenic systemic fibrosis
■Radiation dermatitis
■Radiation dermatitis
■Systemic sclerosis
■Systemic sclerosis
skin reactions to chemotherapy. Chemotherapeutic agents commonly associated with toxic erythema of chemotherapy include cytarabine, anthracycline, 5-FU, capecitabine, taxanes, and methotrexate.46,87 Another differential diagnosis to consider is eruption of lymphocyte recovery, which represents immune system recovery approximately 3 weeks after chemotherapy, and may be associated with fever. Viral reactivation is common post-HCT, including human herpesvirus-6 reactivation, cytomegalovirus, and Epstein-Barr virus, which may present as morbilliform eruptions. As the presentation of chronic GVHD is quite varied, there are many other potential differential diagnoses. An important consideration is that not every cutaneous finding in the post-HCT setting represents GVHD. Biopsy can be useful in helping to distinguish nondiagnostic presentations. One important differential diagnosis in these patients is Demodex folliculitis, which increasingly occurs in the immunosuppressed setting in HCT recipients. Table 129-7 outlines the differential diagnosis of graft-versus-host disease.
COMPLICATIONS
Skin erosions and ulceration caused by chronic GVHD may lead to secondary infection. Wound-healing in these patients is often slow and difficult in the setting of systemic corticosteroids and certain GVHD therapies (eg, sirolimus).88 Sclerotic changes resulting in restriction in joint function may lead to functional disability and joint contractures. Restrictive lung disease may result from sclerotic involvement of the torso.
2331
21
HCT survivors are at increased risk for melanoma and nonmelanoma skin cancer because of previous exposure to ionizing radiation, GVHD-associated immune dysregulation, and immunosuppressive treatment of GVHD.89-91 Both cutaneous squamous cell carcinoma and melanoma also may be increased by long-term treatment with voriconazole, a potent photosensitizer, which may be employed for antifungal treatment or prophylaxis.92,93 Multiple squamous cell carcinomas also have been reported after psoralen and ultraviolet A (PUVA) therapy for GVHD.94 Treatment of skin cancers in these patients is challenging because of the increased risk of infection and poor wound healing.
PROGNOSIS AND CLINICAL COURSE
Although the presence of GVHD is associated with a decreased risk of malignancy relapse, GVHD leads to significant morbidity, especially in patients with steroid-refractory disease. Chronic GVHD is the second leading cause of long-term mortality in HCT patients after recurrence of primary disease.95 Extensive (>50%) skin involvement96 and “lichenoid” skin histology also may be associated with poorer survival. The association between sclerotic skin and mortality is thought to relate to higher doses of immunosuppression required, and therefore increased risk of infection. Sclerotic skin is likely also a marker of more-severe disease, such as lung disease, which may directly impact mortality. A number of systemic risk factors portend a poor prognosis, including a history of progressive involvement from acute to chronic GVHD,97 thrombocytopenia (fewer than 100,000 cells/mL),56 elevated bilirubin,98
older age, GI symptoms, and lack of response to therapy at 6 months.97
PREVENTION
GVHD prevention begins prior to transplantation with selection of the most closely HLA-matched donor, the GVHD prophylaxis regimen, and, in some cases, T-cell depletion or manipulation of the graft. T-cell depletion is accomplished through ex vivo T-cell–negative selection or enrichment of the CD34+ stem cell population, or more commonly through in vivo treatment with anti–T-cell therapy, such as posttransplantation cyclophosphamide,99 or alemtuzumab, an anti-CD52 monoclonal antibody.100,101 Other strategies for T-cell modulation, including IL-2 and antibodies targeting IL-17 and IL-21. have been used to attempt to induce the expansion of T-regulatory cells.102,103 Even though T-cell modulation may lead to higher rates of graft failure, cancer relapse, and infection, studies show that strategies aimed at reducing the incidence of GVHD may lead to improved overall mortality.104,105 Prophylactic immunosuppressive therapy is initiated concomitantly with the administration of the hematopoietic graft, but, as with T-cell depletion, such therapy must
2332
be balanced with the potential for diminished graftversus-leukemia/lymphoma effect and long-term infection risk. One of the most important strategies has been increasing use of antihuman T-lymphocyte immune globulin (or antithymocyte globulin) as part of the conditioning regimen,106,107 which has shown efficacy in acute and chronic GVHD. Administration of antithymocyte globulin in this setting has been shown to significantly reduce the incidence of chronic GVHD at 2 years, and lead to discontinuation of immunosuppression with a trend toward improved survival.108
Sirolimus is an mammalian target of rapamycin inhibitor that has been used in prophylactic regimens in combination with standard therapy or in place of calcineurin inhibitors, and which may have the advantage of improved tolerance compared to calcineurin inhibitors.109,110
Similar to solid-organ transplantation, skin cancer screening and patient education regarding photoprotective measures is a key preventive strategy in all HCT patients, especially those who develop chronic GVHD.111 Patients are also at elevated risk of systemic infection; consequently, implementation of preventive infectious disease recommendations and careful monitoring for cutaneous infection, particularly in patients with chronic skin erosions/ulcerations, is prudent. Finally, patient education regarding early signs of skin sclerosis and fascial involvement, including skin tightness, edema, muscle cramping, and range-of-motion restriction, may facilitate early diagnosis and initiation of treatment.
TREATMENT
MANAGEMENT OF ACUTE GRAFT-VERSUS-HOST DISEASE
MANAGEMENT OF ACUTE
GRAFT-VERSUS-HOST
DISEASE
Treatment of acute GVHD is usually initiated in the hospital, given the proximity to the date of HCT and the need for close observation. Patients with mild (grade I) skin involvement without hepatic or GI symptoms may respond to high-potency topical steroids. However, more-severe skin involvement or the presence of internal organ involvement necessitates treatment with systemic corticosteroids. Patients with moderate-to-severe acute GVHD may warrant treatment with methylprednisone 1 to 2 mg/kg/day; however, lower doses may be adequate to control disease. Patients with skin sloughing require meticulous skin care, infection surveillance, and fluid management similar to toxic epidermal necrolysis. Approximately 50% of patients respond to systemic corticosteroids; however those who require additional therapy typically receive increased doses of their GVHD prophylaxis (most commonly tacrolimus, cyclosporine, or methotrexate) or additional therapy, including horse antithymocyte globulin, extracorporeal photopheresis (ECP), and mycophenolate mofetil.112 Mycophenolate
mofetil is particularly helpful in patients with acute GVHD limited to the skin, with a large case series demonstrating a 92.3% response.113 Other agents include alemtuzumab,100 basiliximab,114 inolimumab,115
daclizumab,116 etanercept,117 infliximab,118 denileukin diftitox, pentostatin,119 sirolimus,120 and mesenchymal stem cells.121 Levine and colleagues117 demonstrated complete remission of skin symptoms in 81% of patients treated with steroids and etanercept, compared to steroids alone (complete remission = 47%). Similarly, infliximab has also shown success in acute treatment-refractory skin GVHD (33% to 60%).118
Immunosuppression to treat acute GVHD must always be weighed against the risk of infection and dampening of the graft-versus-leukemia effect. Administration of ECP is sometimes preferred prior to increased immunosuppression. ECP is thought to modulate alloreactive T-cell and dendritic cell activity. During ECP, the white cell compartment of the blood is removed from the patient via pheresis, mixed with 8-methyoxypsoralen, irradiated with UVA light, and then returned to the patient. In a pooled analysis of studies examining ECP, the overall response rate was 84% for acute skin GVHD.122 In a randomized trial, the addition of ECP to systemic steroids resulted in higher response rates, particularly for skin-only acute GVHD.123 Limitations to ECP include the time commitment for the procedure—requiring several hours on 2 consecutive days for 1 cycle, the requirement and cost of a dedicated pheresis center, potentially prolonged vascular access, and fluid imbalance. The optimal frequency and duration of ECP treatment is unclear. Typically, 1 cycle is given weekly, followed by tapering as a response is achieved. Phototherapy (UVA1,124 PUVA,125 narrow band UVB126) also has been used for acute GVHD,127 but is logistically challenging in the inpatient setting and should be administered cautiously to avoid inducing erythema. In a 2008 study, 39 (71%) of 55 participants with steroid-resistant acute GVHD sustained a complete or partial response to mesenchymal stem cell infusion.128 Responses were seen regardless of mesenchymal stem cell source (HLA-matched, haploidentical, or third-party unmatched donors), and immunogenicity was not observed. In a Phase I study, response rates of 67.5% were reported, with 27.5% experiencing a complete response.121 The immunomodulatory mechanism of mesenchymal stem cells is unclear in GVHD, but may be through the induction of T-regulatory cells.
MANAGEMENT OF CHRONIC GRAFT- VERSUS-HOST DISEASE
MANAGEMENT OF
CHRONIC GRAFT-
VERSUS-HOST DISEASE
The dermatologist should play a key role in the multidisciplinary approach to chronic GVHD management, beginning with careful assessment of the subtype and
21
extent of skin involvement. Together with an understanding of other organ system activity, infection risk, relapse risk, and GVHD prognostic risk factors, a decision regarding the appropriateness of topical, physical (eg, phototherapy), and systemic therapy can then be made. If systemic therapy is prescribed by the transplant physician, periodic dermatologic monitoring is advised to differentiate adverse drug reactions or other new skin disease from GVHD, to assess cutaneous disease response, and to monitor for infection and skin malignancy. For mild chronic skin GVHD without sclerotic features, including lichen planus–like, ichthyotic, and papulosquamous types, topical steroids may be used either alone or in conjunction with systemic steroids with close followup and screening for more significant or systemic disease.129 Treatment typically includes mid- to high-potency topical steroids with topical calcineurin inhibitors130 applied twice daily, potentially under occlusion for thicker or refractory lesions. Topical calcineurin inhibitors are particularly useful for treatment of areas at high risk of skin atrophy, such as the face (including the lips) and intertriginous surfaces. Extensive application of topical calcineurin inhibitors, however, can lead to systemic absorption.131 Additional objectives of topical therapy include relief of pruritus and prevention of skin breakdown and infection. Regular moisturization to maintain skin integrity and strict photoprotection should also be employed. Pruritus without rash can be treated with low-potency topical steroids and menthol-based creams. Although systemic antihistamines at higher-than-standard dosing are often helpful, caution should be taken to avoid inducing or exacerbating oral and ocular sicca symptoms.132
Phototherapy may be appropriate for patients with limited epidermal or sclerotic disease in whom systemic therapy is not otherwise warranted (eg, without internal organ system involvement), or in whom systemic immunosuppressive therapy is contraindicated (eg, active infection). Data are limited to anecdotal cases and case series. Longer-wavelength UVA (340 to 400 nm; UVA-1) may be particularly beneficial for fibrosing forms of chronic GVHD.124 UVA-1 does not require psoralen ingestion/topical, and penetrates deeper into the dermis than full-spectrum UVA. UVA-1 is thought to increase the synthesis of matrix metalloproteinases, and decrease the synthesis of procollagen though IL-1 and IL-6. It may also reduce levels of transforming growth factor-β, TNF, IL-8, and IL-10. Although UVA-1 is not yet widely available in the Unites States, it appears to be well tolerated, acceptable for pediatric use, and is not associated with persistent photosensitivity, carcinogenic risk, or potential GI issues that may occur with oral psoralen use. The largest series describes skin softening following UVA-1 treatment of sclerotic-type GVHD in 17 (70.8%) of 24 GVHD patients, with the best response rate in patients receiving high-dose UVA-1.133 In a study of mediumdose UVA-1 phototherapy in 7 patients with lichenoid GVHD and 3 with sclerotic GVHD, all 3 patients with sclerotic GVHD demonstrated partial response or
2333
21
improvement.134 Ständer and colleagues135 described softening of skin lesions, improved joint mobility, and healing of skin erosions in 5 adult patients with medium-dose UVA-1 and 1 child treated with low-dose UVA-1. Calzavara Pinton and colleagues136 described 5 patients with sclerotic involvement treated with UVA-1 therapy with complete resolution in 3 patients and partial response in 2 patients. Although UVA-1 may accentuate pigmentary abnormalities, the risk of skin cancer is significantly less than the risk associated with PUVA.137 PUVA has shown limited efficacy in treating GVHD and is associated with potential phototoxicity and risk of skin cancer.138 Narrow band UVB also has been used, with some response in patients with primarily “lichenoid” disease.126,139 Care must be taken in administering phototherapy, as UV radiation may trigger flares of cutaneous disease. Skin cancer risk assessment and concurrent use of photosensitizing medications should also be considered. Voriconazole therapy, in particular, is an important photosensitizer in this setting and may increase the risk of squamous cell carcinoma and melanoma.92,93
First-line treatment of moderate to severe GVHD is high-dose systemic steroids, usually in combination with other immunosuppressive agents.129 Long-term systemic therapy is often required for patients with moderate-to-severe disease (3 or more organs involved, moderate or severe involvement in any organ, or lung involvement).50 Combination therapy with azathioprine, thalidomide, mycophenolate mofetil, Plaquenil, and sirolimus have suggested no significant benefit over prednisone alone for first-line therapy.129 Calcineurin inhibitors should be used in combination with systemic steroids, and can be particularly helpful in moderating steroid dependence.140
Among the myriad of systemic treatments that have been used in patients with chronic GVHD who cannot be tapered from systemic corticosteroids or who are steroid-refractory, no single treatment has demonstrated proven superiority. There are no U.S. Food and Drug Administration–approved treatments for chronic GVHD. Determination of a preferred secondline or salvage agent has been complicated by poor understanding of the disease process and a lack of high-quality clinical trials. The need to spur clinical trial development in the field of chronic GVHD was acknowledged by the chronic GVHD NIH Consensus Project, which included a standardized system of organ system assessments and recommendations for clinical trial design.50 Unfortunately, validated measures of cutaneous disease activity still have limitations, and are not routinely implemented in clinical care.141 Ideally, dermatologic collaboration in future therapeutic trials will permit better quantification of cutaneous disease response. Response should be assessed 8 to 12 weeks after initiation of a systemic treatment. If patients progress after 4 weeks, a new treatment option should be offered.129 Significant sclerotic disease may require substantially longer treatment periods to determine an effect. ECP is a major salvage therapy in steroid-refractory chronic GVHD, especially in patients with evidence
2334
of skin sclerosis at high risk of adverse effects from systemic immunosuppression. ECP has an excellent safety profile, but is limiting in that it requires sustained venous access. A pooled analysis of ECP studies for chronic GVHD found an overall response rate of 71% for cutaneous disease.122 In a large retrospective analysis of 71 patients, response was seen in 61% with best response was seen in 44 (61%) of patients, most notably in the skin. Thrombocytopenia was associated with a lower response rate.142 Patients with classic chronic GVHD and overlap acute and chronic GVHD demonstrated superior survival after treatment with ECP.143 ECP may be particularly useful for patients with deep-seated sclerotic involvement of the subcutaneous tissue and fascia. Although GVHD-related fasciitis resembles eosinophilic fasciitis, in contrast to eosinophilic fasciitis, GVHD-related fasciitis does not respond well to steroid therapy and may result in significant long-term functional disability. Several case reports describe successful use of ECP for GVHDrelated fasciitis.144
Imatinib mesylate, a multikinase inhibitor with activity against bc-abl, c-kit, PDGFR, and other kinases, has been reported to have a modest benefit at doses lower than are typically administered for treatment of chronic myeloid leukemia (adult: 400 mg daily; children 260 mg/m2 daily) as the drug in GVHD patients is poorly tolerated. In a prospective study specifically for sclerotic skin GVHD, Baird and colleagues145 observed a partial response in 5 of 14 patients, with an average range-of-motion improvement of 26%. The drug is generally well tolerated in the setting of treatment of chronic myelogenous leukemia; however, side effects are common in the GVHD setting and include hypophosphatemia, fatigue, nausea, diarrhea, edema, and muscle cramping. The modest effect reported is in line with a large series reporting limited efficacy,146 despite many prior case series and other studies revealing more promising results.147-149 Ibrutinib, a small-molecule inhibitor of Bruton tyrosine kinase, has shown preclinical efficacy in mouse models of GVHD, and clinical trials are ongoing.150-152
Rituximab has immunoregulatory effects in patients with chronic GVHD, and data suggesting elevated levels of BAFF in chronic GVHD patients supports this.32 In a prospective, multicenter randomized trial, a significant clinical response in skin sclerosis and joint range of motion was seen in 27% of patients receiving rituximab, compared to 36% of patients receiving imatinib. Among rituximab patients, those with higher circulating activated B cells were more likely to have treatment success.35
Inhibitors of Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling, ruxolitinib and tofacitinib, have also shown promising preclinical and early clinical results.153,154 One study reported an 81.5% response rate in 54 patients with acute GVHD and 84.4% response rate for 41 patients with chronic GVHD treated with ruxolitinib. Cytopenias and cytomegalovirus reactivation were observed during treatment with ruxolitinib.153
Immunosuppressive Agents
■Mycophenolate mofetil156
■Calcineurin inhibitors (cyclosporine, tacrolimus)157
Chemotherapeutic Agents
■Thalidomide158
■Azathioprine159
■Methotrexate160
■Pentostatin161
Antimicrobial Agents
■Cyclophosphamide162
■Clofazimine163
■Hydroxychloroquine132
Small Molecule Inhibitors
■Imatinib,145 dasatinib, nilotinib
■Sirolimus155
■Ruxolitinib,153 tofacitinib154
■Vorinostat164
■Erlotinib165
Antibodies and Fusion Proteins
■Antithymocyte globulin108
■Rituximab35
■Infliximab, etanercept166
■Alefacept167
■Denileukin diftitox, basiliximab, daclizumab116
Cytokines
■Low-dose interleukin-2168
Cellular Therapies
■T-reg adoptive therapy169
■Mesenchymal stem cell therapy170
Radiation
■Total nodal irradiation171
■Thoracoabdominal irradiation172
Other
Other
■Retinoids173
■Retinoids173
■Intravenous immunoglobulin174
■Intravenous immunoglobulin174
■Extracorporeal photophoresis122
■Extracorporeal photophoresis122
■Bortezomib175
■Bortezomib175
■Vismodegib176
■Vismodegib176
Sirolimus also has been used therapeutically in chronic GVHD, with a Phase II trial demonstrating clinical response in 15 (79%) of 19 patients.155
SUPPORTIVE CARE
SUPPORTIVE CARE
Supportive care including physical and occupational therapy is especially important in patients with skin sclerosis and fasciitis leading to joint contractures. Weight-bearing exercise for 30 minutes daily 5 days per week are important for preserving range of motion and bone health. Deep-tissue massage may be helpful to preserve range of motion in patients with fasciitis.132
21
TREATMENT OF CHRONIC ORAL AND VULVOVAGINAL DISEASE
TREATMENT OF CHRONIC
ORAL AND VULVOVAGINAL
DISEASE
Limited oral mucosal disease can be controlled with application of a high-potency topical corticosteroid gel or paste (fluocinonide gel 0.05%, clobestasol gel 0.05%, triamcinolone 0.1% dental paste). Solutions of dexamethasone 0.5 mg/mL and prednisolone 15 mg/mL also may be used as rinses, and are beneficial for widespread involvement and should be swished in the mouth for 4 to 6 minutes 4 to 6 times daily.177 Refractory lesions may respond to intralesional triamcinolone injection. Topical application of tacrolimus 0.1% ointment also may be used in conjunction with steroids and for maintenance; however, systemic absorption has been reported. Calcineurin inhibitors also can be constituted as oral solutions. Topical antiinflammatories should be used in combination with an antiyeast wash (nystatin) as to avoid secondary Candida infection in the setting of oral steroids. Cyclosporine and azathioprine rinses also may be used for refractory disease, but require pharmacy compounding. Patients with salivary gland disease should avoid oral antihistamines as well as other xerogenic medications (selective serotonin reuptake inhibitors, tricyclic antidepressants). Dental hygiene is very important in patients with decreased salivary function and home fluoride treatment is frequently recommended. Salivary stimulants (eg, sugar-free gum) and sialogogue therapy (cevimeline, pilocarpine) are recommended for patients with severe salivary gland dysfunction.132
Although sclerotic involvement of perioral skin involvement is uncommon, in this setting aggressive systemic therapy is indicated. Genital erosions and fissures associated with chronic vulvovaginal disease may be treated with clobetasol propionate ointment nightly, which should be tapered to a maintenance level of 2 to 3 times weekly. Fluocinolone 0.025% ointment can be used in cases of mild disease or for maintenance in cases of treated severe disease. Additionally, tacrolimus ointment has been commonly employed for maintenance therapy in this area. If estrogen is not contraindicated, hormone replacement via topical cream, vaginal ring, or oral replacement may improve genital skin integrity. Limited vaginal scarring/synechiae can be treated with dilators or manual lysing; however, thick vaginal scarring may require surgical intervention.178
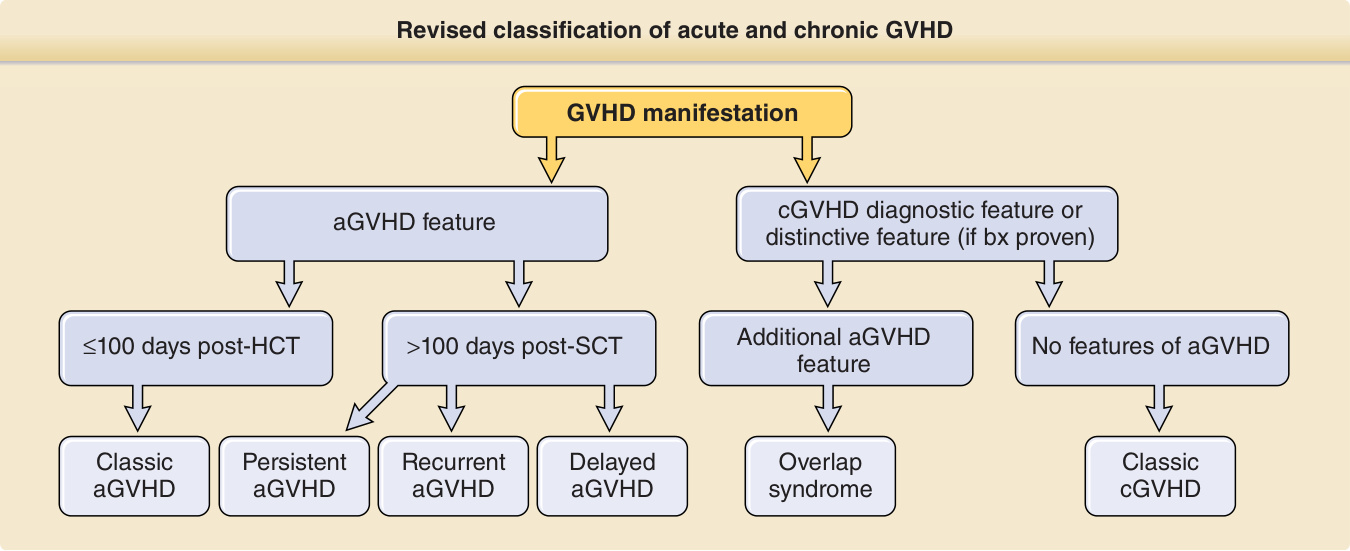
Figure 129-1 Revised classification of acute and chronic graft-versus-host disease (GVHD). aGVHD, acute graft-versushost disease; bx, biopsy; cGVHD, chronic graft-versus-host disease; HCT, hematopoietic cell transplantation; SCT, stem cell transplantation. (Adapted from Jagasia MH, Greinix HT, Arora M, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant. 2015;21:389-401.e1.)
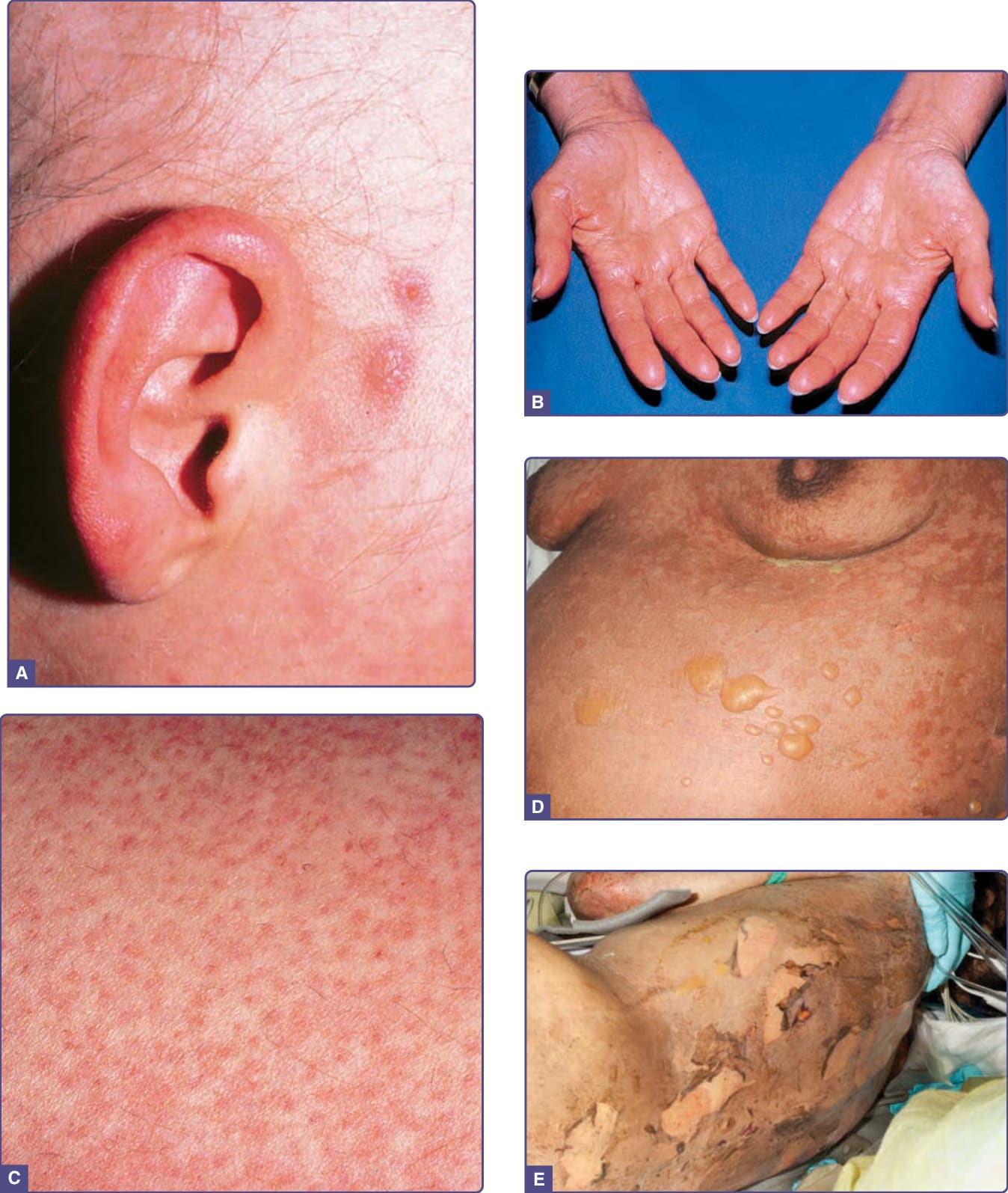
Figure 129-2 Spectrum of acute graft-versus-host skin manifestations. Acute cutaneous graft-versus-host reaction. Erythematous macules involving the ears (A), palms (B), and soles are characteristic of early cutaneous involvement. C, Follicular graft-versus-host disease (GVHD). Perifollicular involvement is an early manifestation of skin involvement. D, GVHD-associated necrolysis. Acute GVHD with bullae formation following donor leukocyte infusion for relapsed acute lymphoblastic leukemia following allogeneic hematopoietic cell transplantation. E, Skin sloughing in a toxic epidermal necrolysis–like presentation of acute GVHD.
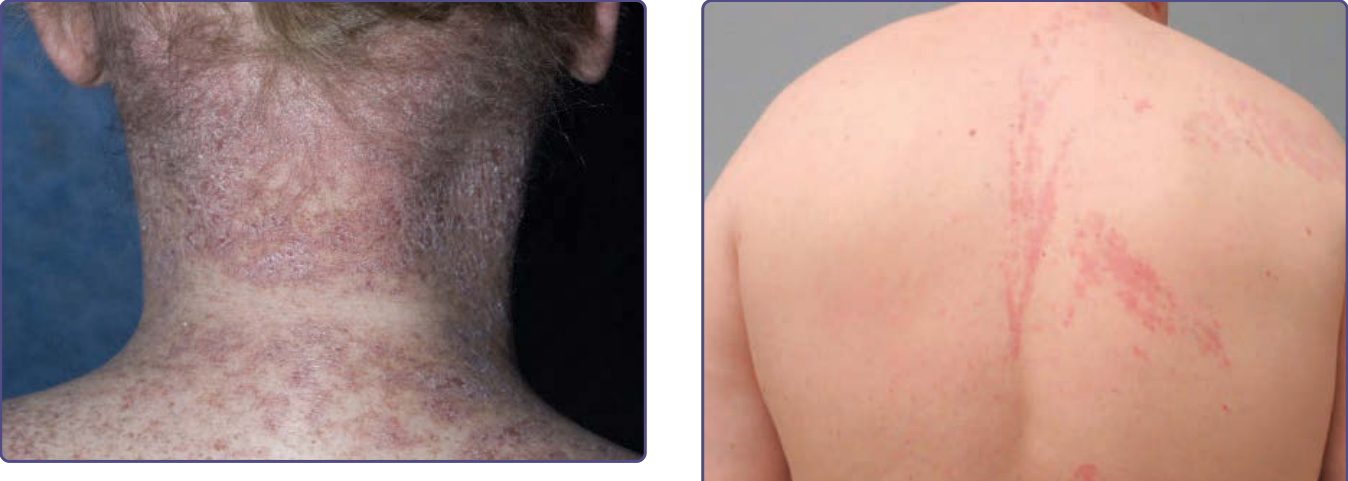
Figure 129-3 Lichen planus-like chronic graft-versus-host disease. Reticulate violaceous plaques with dry scale on the posterior neck and upper back.
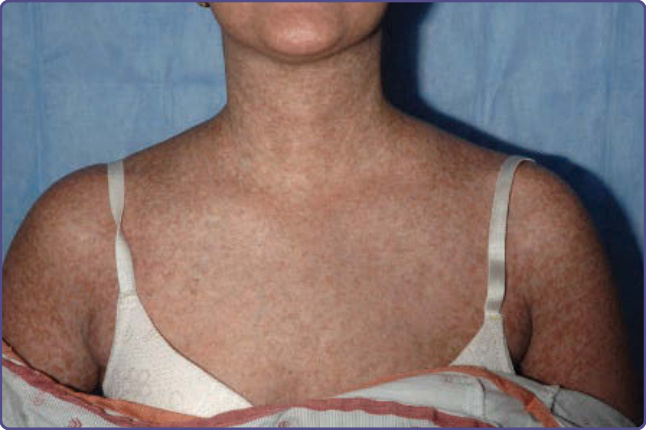
Figure 129-4 Poikilodermic chronic graft-versus-host disease. Hypopigmentation, hyperpigmentation, and erythema on the chest and proximal arms.
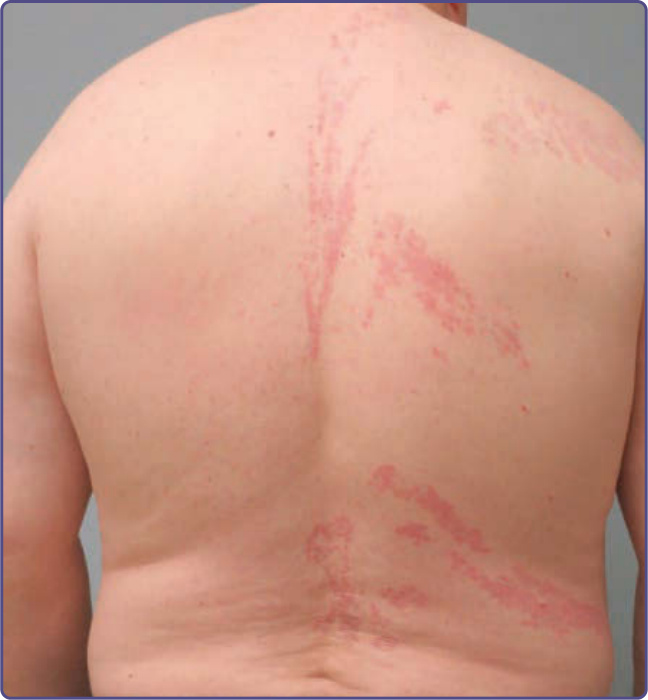
Figure 129-5 Blaschkoid chronic graft-versus-host disease (GVHD). Lichen planus–like chronic GVHD presenting unilaterally in a Blaschkoid distribution of the right-side of the back. (Used with permission from Milan J. Anadkat, MD.)
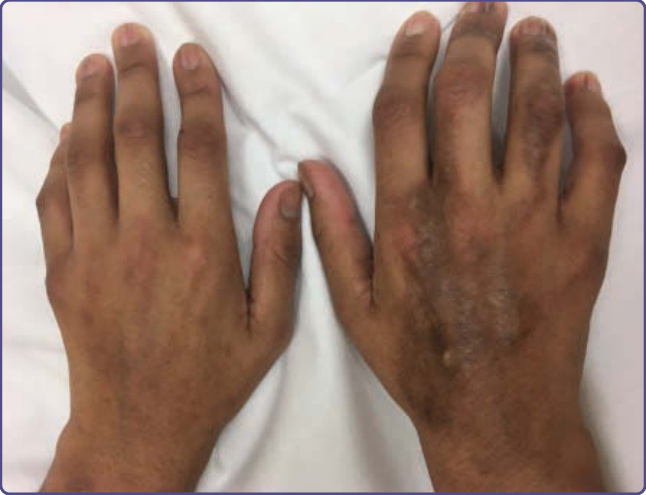
Figure 129-6 Morphea-like hyperpigmented, shiny sclerotic plaques of the dorsal hands and fingers causing functional disability.
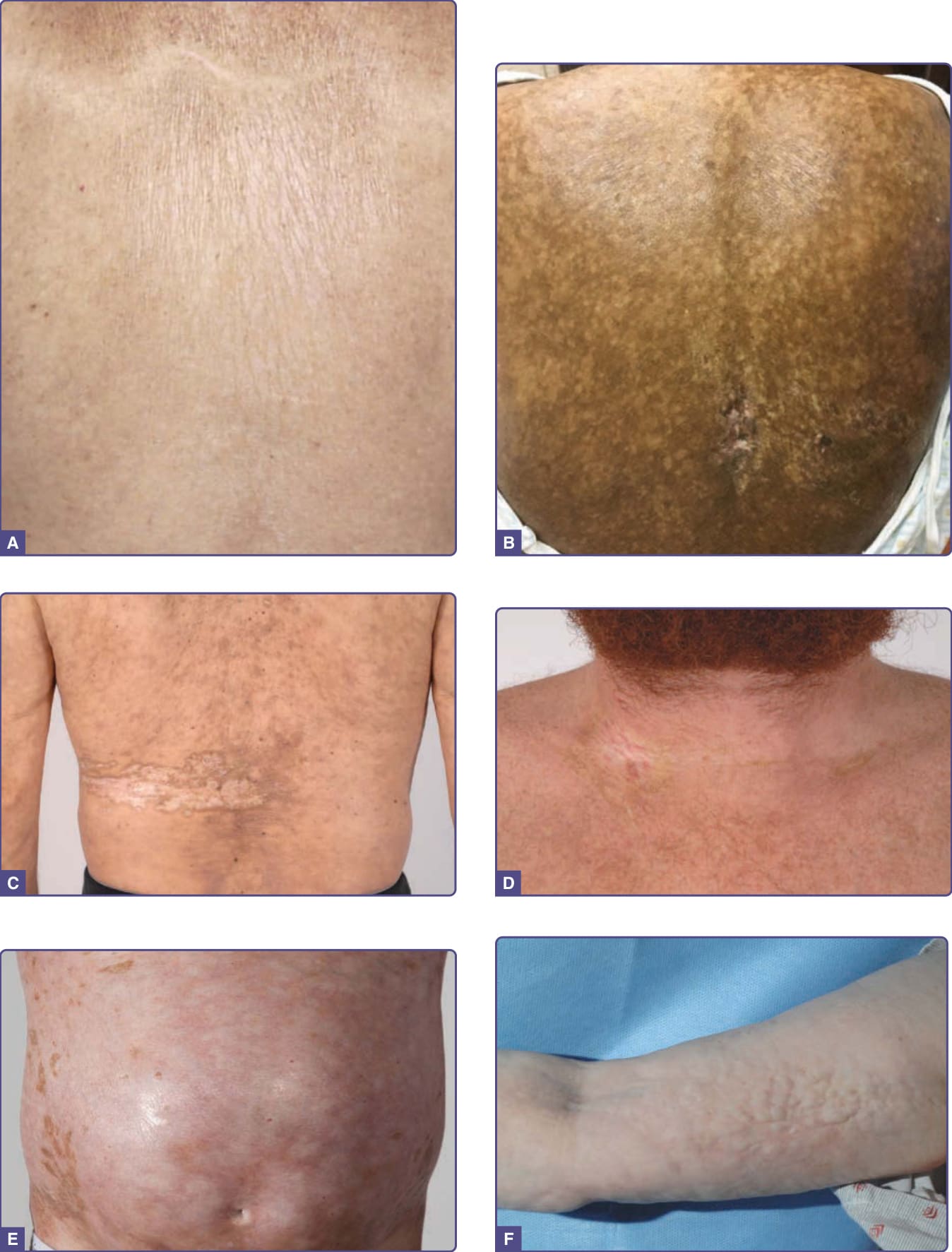
Figure 129-7 Clinical spectrum of sclerotic graft-versus-host disease (GVHD) skin manifestations. A, Guttate white plaques on the upper back resembling lichen sclerosus. B, Hypopigmentation and hyperpigmentation with extensive superficial and deep dermal sclerosis on the back. C, Morphea-like sclerotic plaques in the distribution of a prior zoster infection at this site of the left back and flank. D, Morphea-like sclerotic plaques at sites of previous indwelling line placement near the clavicle (isotopic response). E, Diffuse dermal sclerosis resembling scleroderma on the anterior torso with patchy hyperpigmentation. F, Subcutaneous fibrosis of chronic GVHD. There is prominent rippling with a firm nodular texture extending along the medial arm resembling eosinophilic fasciitis. There is associated decreased range of motion at the elbow.
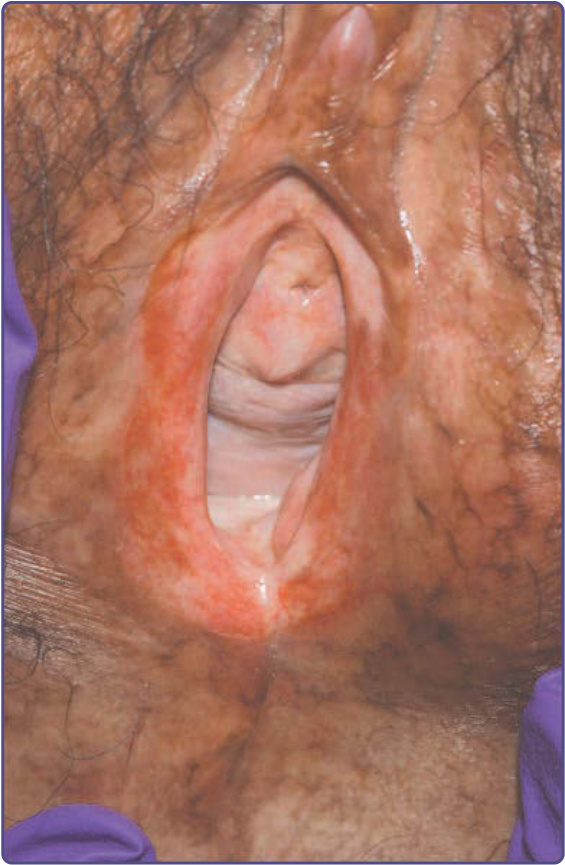
Figure 129-8 Severe chronic graft-versus-host disease (GVHD) of the vulva. The labia minora are partially resorbed with residual vulvitis and atrophic mucosa. Surrounding reticulate hyperpigmentation of the nonmucosal skin is consistent with postinflammatory changes of chronic GVHD.
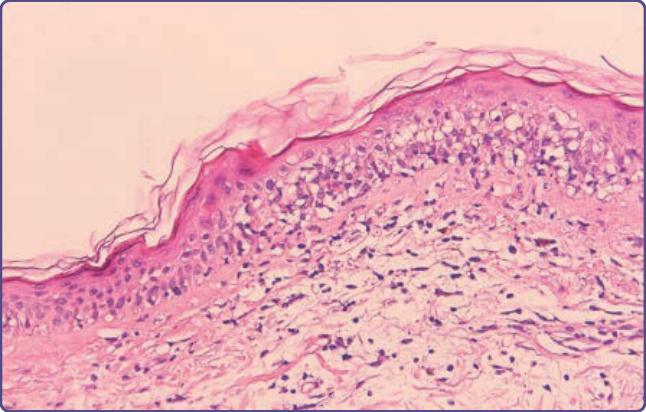
Figure 129-9 Histopathologic features of acute cutaneous graft-versus-host disease, grade II. Inflammation of the upper dermis is present, with extension of lymphocytes into the dermis and interface change.
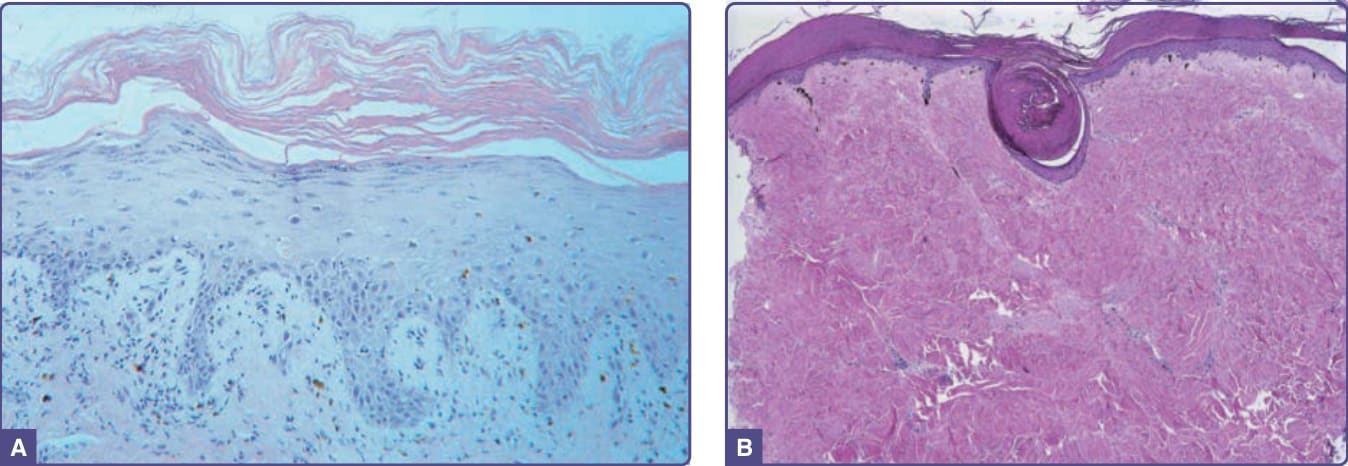
Figure 129-10 Histologic features of epidermal and sclerotic-type chronic cutaneous graft-versus-host disease. A, Histopathologic features of a lichen planus–like reaction. Acanthosis, hypergranulosis, hyperkeratosis, and pointed rete ridges are present. The inflammatory infiltrate is less dense than that usually seen in idiopathic lichen planus. B, Sclerotic-type graft-versus-host disease. There is mild, compact hyperkeratosis of the epidermis with keratin plugging. There is hyalinization of the collagen throughout the dermis with loss of appendageal structures.
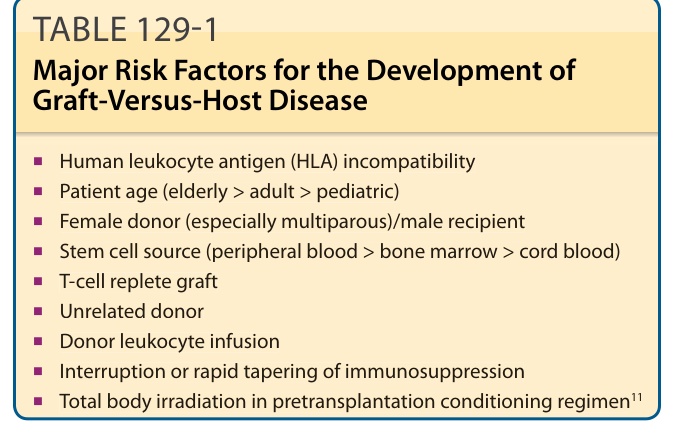
TABLE 129-1 Major Risk Factors for the Development of Graft-Versus-Host Disease
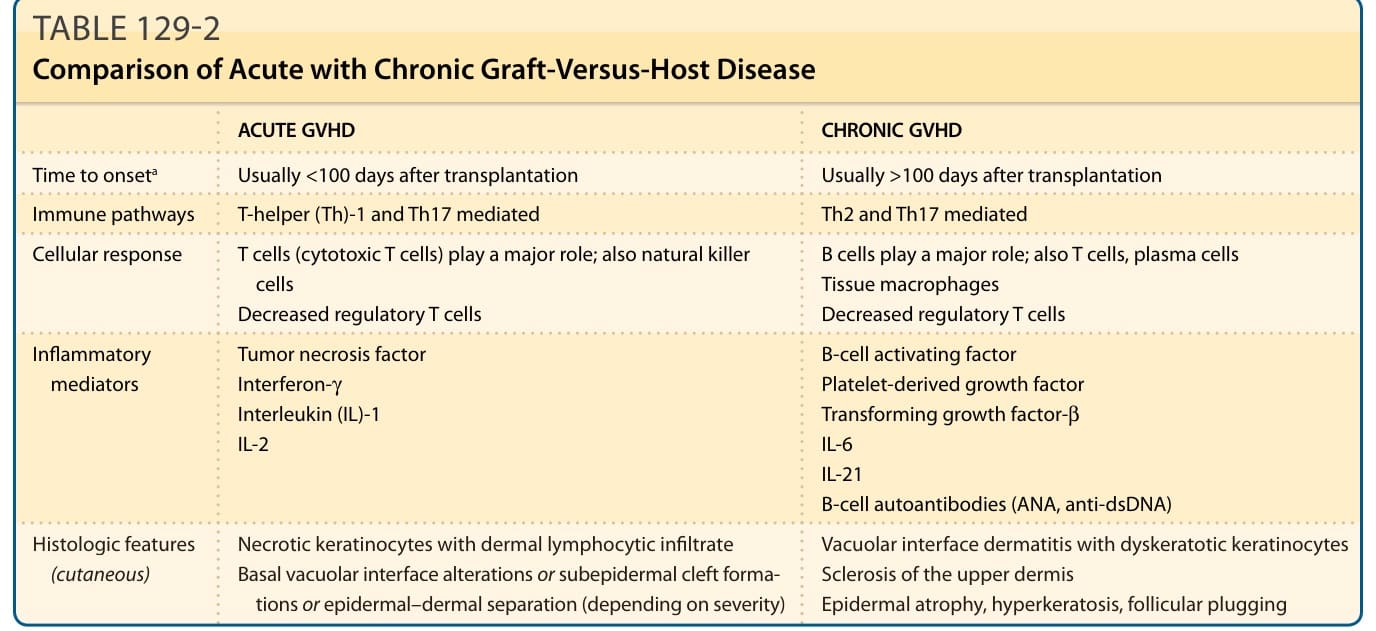
Table 129-2 compares acute GVHD with chronic GVHD. The fundamental concept of GVHD is encompassed by the balance between immunosuppression, induced by the conditioning regimen and GVHD prophylaxis, and immune system (graft) activation. In 1966, Billingham proposed 3 basic requirements for GVHD: (a) immunocompetent transplanted cells, (b) host antigens recognizable by the transplanted cells and lacking in the donor, and (c) a host incapable of mounting an immune response to the transplanted cells.15 The immunocompetent cells are T cells, targeting HLAs expressed on host tissues, as well as key
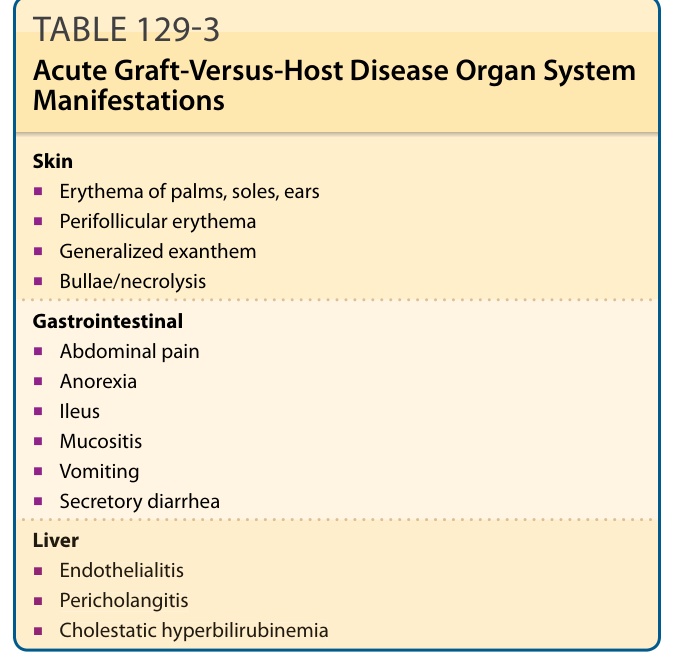
TABLE 129-3 Acute Graft-Versus-Host Disease Organ System Manifestations
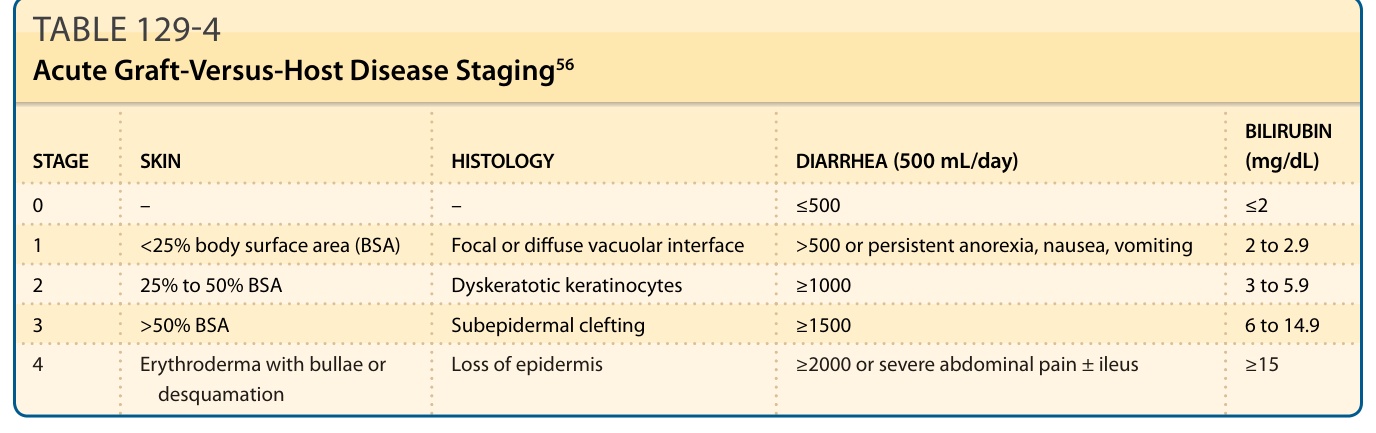
TABLE 129-4 Acute Graft-Versus-Host Disease Staging56
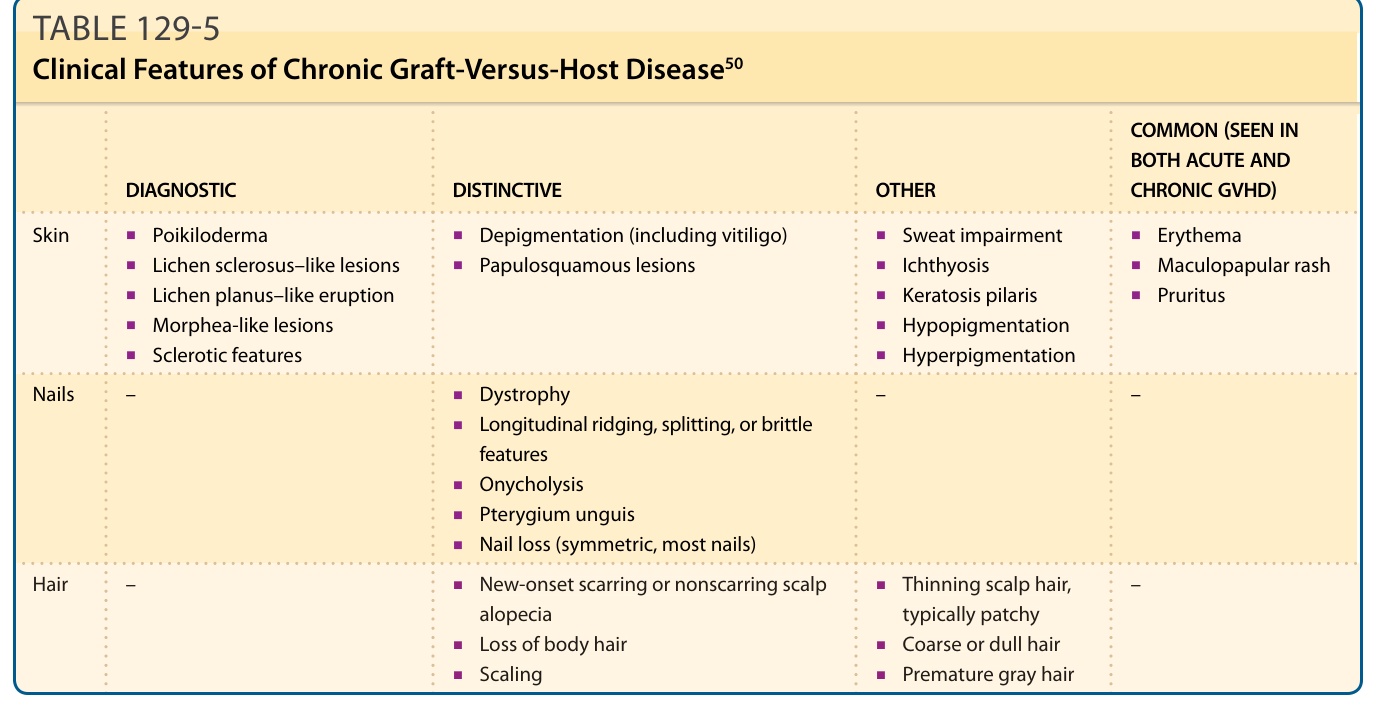
TABLE 129-5 Clinical Features of Chronic Graft-Versus-Host Disease50
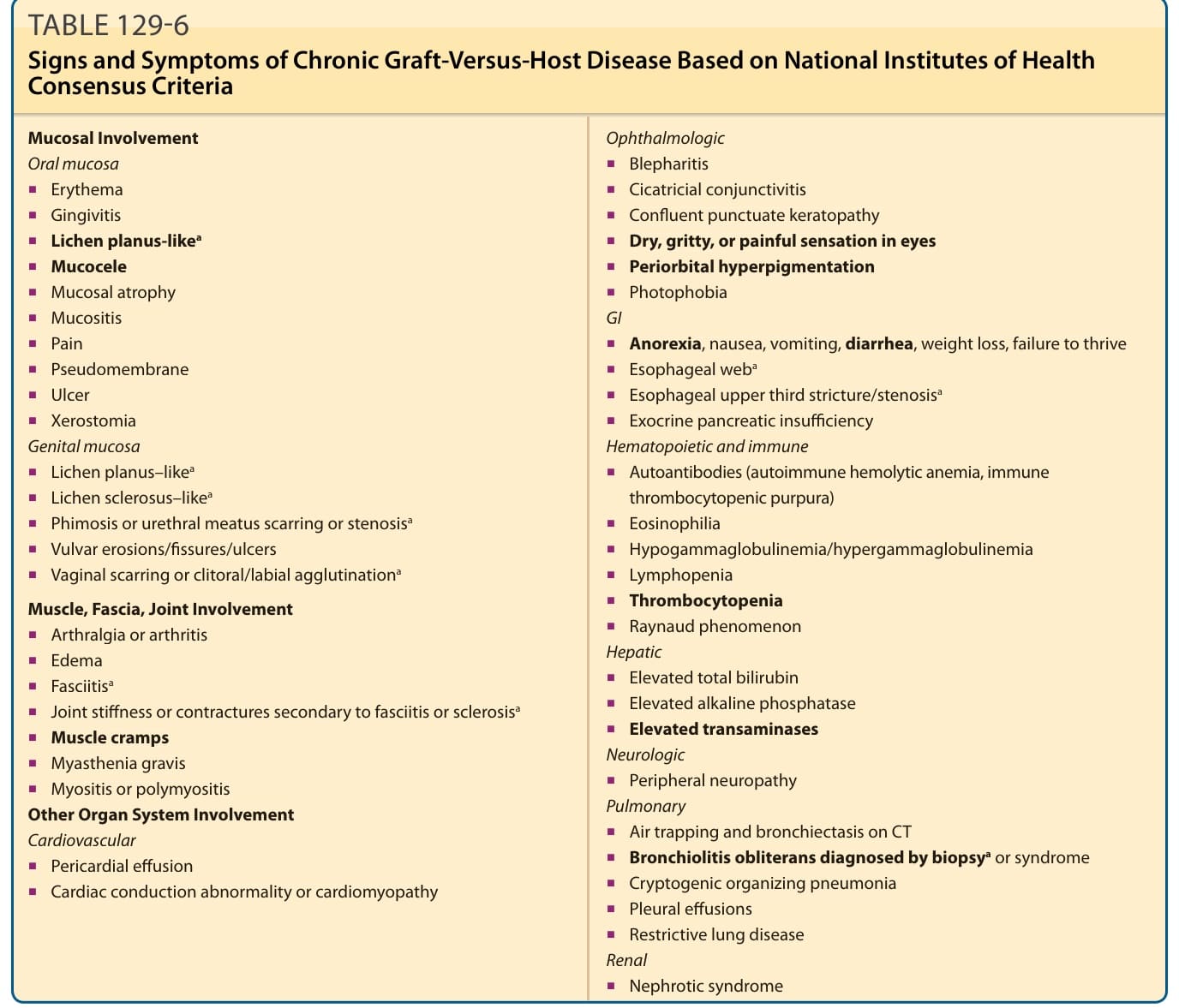
TABLE 129-6 Signs and Symptoms of Chronic Graft-Versus-Host Disease Based on National Institutes of Health Consensus Criteria
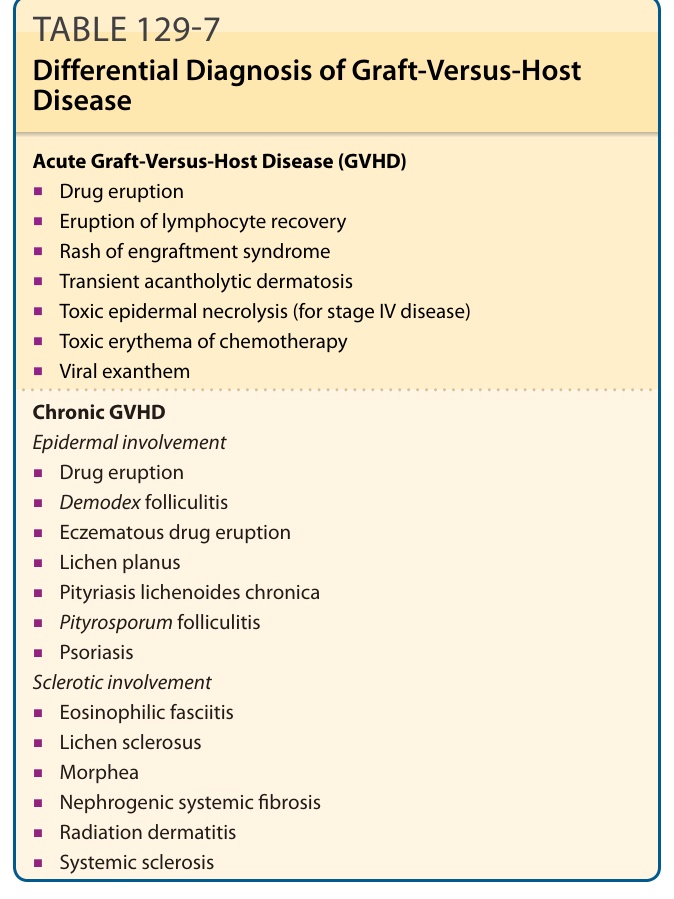
TABLE 129-7 Differential Diagnosis of Graft-Versus-Host Disease
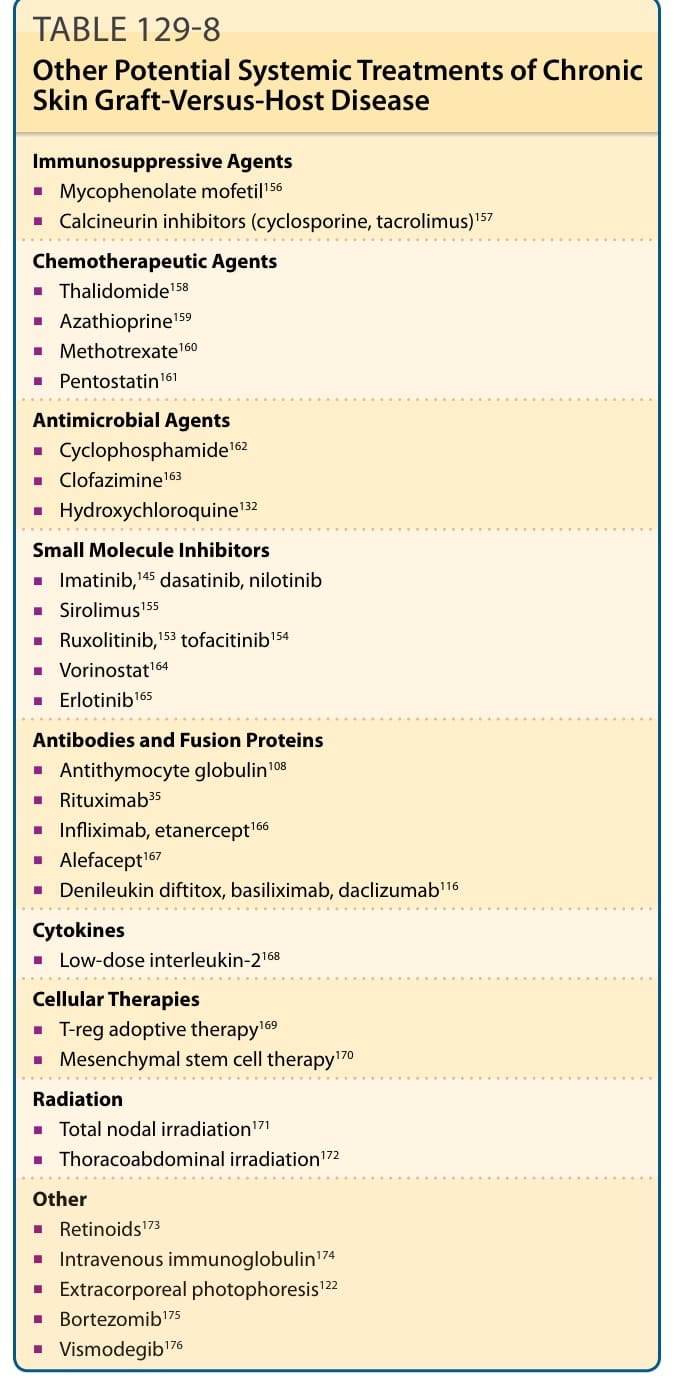
TABLE 129-8 Other Potential Systemic Treatments of Chronic Skin Graft-Versus-Host Disease