Amyloidosis
21
AT-A-GLANCE
■ There are 36 types of amyloid disease, defined by the subunit proteins that constitute the fibrils in these disorders, 11 of which involve the skin.
■ Cutaneous amyloidosis may reflect a systemic form of amyloid, or be localized to the skin and/or mucous membranes.
■ Precursors of fibril subunit proteins may circulate in blood and/or be synthesized locally; they may be wildtype or mutant molecules that can be typed by DNA sequencing.
■ Amyloid in skin may be sampled by lesional biopsy or abdominal fat pad aspiration or biopsy; it is characterized by metachromasia and applegreen birefringence after staining with Congo red, or yellow-green fluorescence after staining with thioflavin T.
■ Deposits may localize to the papillary dermis (macular and lichen amyloidosis) or be characterized by congophilic angiopathy and involvement of adnexal structures (nodular amyloidosis).
■ Amyloid can be typed by immunohistochemistry, immunoelectron microscopy, and/or laser capture mass spectroscopy followed by protein sequencing.
■ Treatment of amyloid diseases is determined by the fibril subunit protein, associated clinical manifestations, and whether disease is systemic or localized.
■ Treatment strategies include suppression of precursor protein, disruption of oligomers and/or fibrils, and/or enhanced clearance of deposits.
■ Localized cutaneous amyloidoses present specific challenges and opportunities for diagnosis and therapy because of the accessibility of skin lesions, association in some cases with defined genetic abnormalities, and multifactorial etiologies for itch in pathogenesis.
DEFINITION
Amyloid is a pathologic entity characterized by the presence of mostly extracellular homogenous hyaline material that is typically metachromatic, as seen by color changes of dyes such as Congo red (actually orange in solution), crystal violet (which yields a deep red-purple color on a blue background), or sodium sulfate Alcian blue (reacts with proteoglycan in amyloid deposits to yield a green color). Other stains that
can be used include cotton dyes such as Sirius red (a polyazo dye) and Pagoda red. Amyloid is further defined in tissue section as yielding apple-green birefringence by polarizing microscopy after staining with Congo red, and as having typical yellow-green fluorescence after staining with thioflavin T. Ultrastructurally, the main constituents of deposits are fibrils which are 10 to 15 nm in diameter, and variable in length, that accumulate in the extracellular space, and can be visualized by electron microscopy. A second feature of all amyloid deposits is the presence of a pentraxin, serum amyloid P-component (SAP). In addition to the fibril and SAP, deposits are enriched in heparan sulfate proteoglycan, as well as a number of proteins that have been identified by extraction of amyloid from tissue. These other substances include several apolipoproteins, notably apoE, apoJ, and apoA4.1 Thus the diagnosis of amyloidosis is rigorously defined by the appearance, tinctorial properties and ultrastructural features of biopsied material (Table 125-1).
AMYLOID DISEASES RELEVANT TO DERMATOLOGY
Different forms of amyloid disease are distinguished by the subunit proteins that constitute the fibril, 36 of which have been described, each associated with distinct clinical entities,2 11 known to involve the skin and/or have cutaneous manifestations (Table 125-2). The current list of 36 subunit proteins includes 6 different apolipoproteins, notably the high-density lipoprotein-associated serum amyloid A (apoSAA), an acute-phase reactant that forms the amyloid in reactive or secondary (AA) amyloidosis, and 2 members of the immunoglobulin gene superfamily, immunoglobulin light chain (AL), the major constituent of primary or myeloma-associated amyloidosis, and β2- microglobulin (Aβ2m), the subunit protein in patients on longstanding hemodialysis or chronic peritoneal dialysis (dialysis amyloidosis). Fibril subunit proteins arise from soluble precursors, most of which can be found in blood, but which also may be synthesized by cells contiguous to the amyloid deposits. They may be wildtype (WT) (ie, have the same amino acid sequence as the physiologic precursor), but in several forms of amyloidosis they have been found to have single nucleotide and amino acid substitutions, corresponding to point mutations in the DNA sequence. These mutations characterize familial forms of amyloidosis, in which multiple members of a kindred are affected by distinct organ-specific clinical syndromes, but may
■Homogenous-hyaline
■Homogenous-hyaline
■Mostly extracellular
■Mostly extracellular
■Typical 10 to 15 nm fibrils by electron microscopy
■Typical 10 to 15 nm fibrils by electron microscopy
■Metachromasia
■Metachromasia
■Congo red birefringence
■Congo red birefringence
■Thioflavin T yellow-green fluorescence
■Thioflavin T yellow-green fluorescence
■Multiple chemical types
■Multiple chemical types
also occur in apparently sporadic cases of amyloidosis. More than 500 different mutations in subunit proteins, or in proteins strongly associated with several forms of amyloidosis, have been identified, providing diagnostic genetic markers for several important forms of amyloid disease.3
PATHOGENESIS
Following synthesis in the cell of origin, precursor proteins may adopt a number of configurations, some of which may be normal folding intermediates important for intracellular processing and secretion. Cellular factors that contribute to protein misfolding include membrane interactions, altered cell chemistry, posttranslational modifications, crowding, and pathogenic mutations; in both the intracellular and extracellular milieu, acidification, temperature, protein concentration, and oxidative stress may be important.4
Prior to the formation of amyloid fibrils, precursor proteins may adopt the configuration of amorphous aggregates, oligomers, and other intermediates, which may be demonstrable in vitro, and in several cases in vivo. Aggregates have not yet accumulated cofactors, such as SAP and heparan sulfate proteoglans,
21
but appear to be important in mediating distinct functional abnormalities that are demonstrable in association with, or preceding, amyloid deposition, as well as inducing apoptosis and cell death. In addition, several precursor proteins undergo proteolysis as part of the conversion to insoluble fibrils, with specific cleavage products being the main form of subunit protein retrieved from deposits. However, the precursor– product relationships of amorphous aggregates, oligomers, and alternative fibrillar states have yet to be fully defined (Fig. 125-1).1,4
SYSTEMIC FORMS OF AMYLOIDOSIS
The majority of patients seen in tertiary care centers have systemic forms of amyloidosis, most commonly AL, AA, and amyloidosis caused by transthyretin (ATTR), a thyroid transport protein also known as prealbumin. In a 2013 series from the United Kingdom, 65% of patients with systemic amyloid were AL, 18% AA, 7% WT ATTR, and 10% mutant ATTR.5 In AL, high levels of a monoclonal immunoglobulin (Ig) light chain are produced by an aberrant plasma cell population, associated with frank or smoldering multiple myeloma in 10% to 20%. Sustained elevated levels of SAA are triggered by proinflammatory cytokines, notably interleukin (IL)-6, which may be a result of chronic infections or rheumatic or autoinflammatory diseases. In ATTR, the conversion of circulating WT or mutant TTR may be reflected in relatively low levels of precursor protein as a result of a “sink” effect in which precursor may deposit onto existing amyloid. Organ system involvement for AL is clinically diverse, with cardiac, renal and neurologic disease being particularly important; in AA amyloidosis, approximately 80% renal involvement occurs, and is the cause of morbidity and mortality, with lesser
FIBRIL PROTEIN PRECURSOR PROTEIN TARGET ORGANS CUTANEOUS INVOLVEMENT
AL Immunoglobulin light chain All except CNS Pinch/postproctoscopic purpura; nodules; waxy plaques; congophilic angiography; macroglossia
Aβ2M β2-Microglobulin Musculoskeletal Subcutaneous and lingual nodules
AA Serum amyloid A All except CNS Fat pad aspiration/biopsy
ATTR Transthyretin Nervous system; heart; vitreous Periorbital purpura; nodules; fat pad aspiration/biopsy
AApoAI Apolipoprotein AI Heart; liver; kidney; nervous system C-terminal variants affect larynx and skin; yellow and maculopapular squamous lesions
ALys Lysozyme Kidney Variants may present with sicca, have adenopathy, petechial rash/bruising
AGel Gelsolin Cornea; peripheral nervous system Cutis laxa; pruritus; ecchymoses; alopecia
ACys Cystatin C Cerebral hemorrhages Asymptomatic
AGal7 Galectin 7 Localized cutaneous Lichen/macular amyloidosis
AIns Insulin Iatrogenic Injection site (insulin pump)
2259
AEnf Enfuvirtide Iatrogenic (HIV medicine) Local injection site
AEnf Enfuvirtide Iatrogenic (HIV medicine) Local injection site
21
Alternative conformations of amyloidogenic proteins
Unfolded
1 3
Folding intermediates
Energy
Partially folded states
Native
Protein misfolding causes:
Posttranslational modifications Oxidative stress Protein concentration Mutations Temperature Altered cell chemistry Crowding Membrance interactions
β-Linked protein and inclusion
Collins body
Neuroserpin
4
Lewy body
α-Synuclein
Variant Creutzfeldt- Jakob disease amyloid
Prion protein
Oligomers
β-Amyloid peptide β-Amyloid plaque
Pick body Tau protein
Glutamine repeats (huntingtin)
Huntingtin inclusion
Amyloid fibrils Amorphous aggregates
Intramolecular contacts
Front Mol Biosci 2016; 3:1
Intermolecular contacts
involvement of the GI tract (20%) and heart; ATTR may present as overlapping syndromes of neuropathy, cardiomyopathy, GI, vitreous, and occasional leptomeningeal amyloidosis.6
Less-common forms of systemic amyloidosis include several heredofamilial syndromes that have been prominently associated with nephropathic amyloidosis (apoA2, apoCII and apoCIII, fibrinogen, lysozyme), and lattice dystrophy, cutis laxa, and cranial neuropathy (gelsolin).3
Although dominant organ involvement may define clinical presentation in each of the systemic amyloidoses, there is often diffuse involvement that may become more apparent as disease progresses, or is apparent at postmortem. Organ-dominant disease may significantly influence the prognosis of patients with AL, with cardiac involvement accounting for a 1-year mortality of approximately 45% and a median survival time of 6 to 14 months compared to a subset of patients with soft-tissue (eg, macroglossia, submandibular swelling), bone, and joint disease, among whom 2- to 3-year survivals are not uncommon. In AA, most patients have glomerular disease, presenting as proteinuria/nephrotic syndrome; however, there also
2260
may be interstitial and vascular amyloid, which may present as an elevation in creatinine, or renal insufficiency, without significant proteinuria. WT and mutant ATTR may present as carpal tunnel syndrome with amyloid retrievable from the flexor retinaculum in cases that have been available for study. Although most mutant ATTR are associated with neuropathy, approximately 50% also have cardiomyopathy, which may dominate the clinical presentation in patients with specific mutations associated with late-onset disease.
SKIN DISEASE IN SYSTEMIC AMYLOIDS
SKIN DISEASE IN SYSTEMIC
AMYLOIDS
All forms of systemic amyloid may be associated with occult or overt cutaneous involvement, again with variable clinical presentations. Overt presentations include pinch and periorbital purpura in approximately 10% of patients with AL (Fig. 125-2) and a smaller number with ATTR. Macroglossia, lingual nodules, or lateral ridging, are unique to AL (Fig. 125-3) and Aβ2m.
Dermatologic signs may be found in as many as onethird of patients with AL amyloid. Almost any site has been described, including face, neck and fingers. Sclerodermoid and bullous lesions may be found rarely7; hair and nail changes can also occur. In AL, fissured masses may be present focally or diffusely in the papillary or reticular dermis. A common and often diagnostic
21
feature is the presence of amyloid in the walls of small or medium-sized blood vessels (Fig. 125-4), causing vascular wall weakening and hemorrhage. Overt cutaneous involvement is rare in AA amyloidosis; however this form of amyloid may complicate inflammatory cutaneous disorders, most notably in patients with severe inflammation complicating Hidradenitis suppurativa, pyoderma gangrenosum/ inflammatory bowel disease, and some patients with severe and uncontrolled psoriatic arthritis. AA amyloidosis may also complicate autoinflammatory diseases that have distinctive skin lesions such as erysipelaslike erythema (familial Mediterranean fever), cold urticaria (some of the cryopyrinopathies), erythematous macules and papules (hyper-IgD syndrome), and migratory edematous patches or periorbital edema (tumor necrosis factor receptor-associated periodic syndrome). Cutaneous manifestations may occasionally be found in the hereditary forms of amyloid. ATTR may be associated with eyelid and peripheral ecchymoses, atrophic scars, bullous lesions, thickened skin, and xerosis8; cutis laxa is a major manifestation of AGel9; yellow and maculopapular squamous rashes may complicate carboxyterminal variants of AApoAI10; and petechial rashes/easy bruising have been described in some variants of ALys11; asymptomatic skin deposition has been found for ALys and ACys12 (see Table 125-2).
EPIDEMIOLOGY OF THE SYSTEMIC AMYLOIDS
The overall incidence of systemic amyloid is 5 to 12 cases per million-person years in large series reported from the Mayo Clinic, the United Kingdom amyloidosis referral center, and Scandinavia.5,13 During the 20th century, the percent contribution of AA amyloid resulting from chronic infection decreased, correlating with an increased representation of associated rheumatologic, inflammatory bowel and autoinflammatory diseases such as familial Mediterranean
2261
21
fever. There are an estimated 5000 to 10,000 cases of AA amyloid in Europe and North America. Approximately 100,000 persons are affected by familial Mediterranean fever worldwide; in Turkey, Syria, Egypt, and Israel, it is a common cause of AA, and is responsible for a significant percent of renal disease caused by amyloidosis. Each year, 1275 to 3200 new cases of AL amyloid are reported, giving an estimate of 15,000 patients in the United States and Europe. The overall incidence of familial amyloid polyneuropathy caused by ATTR is 0.3 cases per year per million persons, or 5000 to 10,000 cases worldwide. However, there are wide variations in incidence for the most common Met30 ATTR mutation, which is endemic in northern Portugal and the north of Sweden. The Ile122 ATTR mutation is carried by 4% of the African American and African Caribbean populations, and in parts of west Africa.3,13,14
LOCALIZED AMYLOIDOSIS
Excluding CNS amyloid associated with neurodegenerative diseases, localized forms of amyloid account for approximately 10% of patients seen in referral centers. They include nasopharyngeal (laryngeal), ocular, genitourinary, and pulmonary amyloidoses, as well as amyloidomas that may occur in a variety of locations. Symptoms reflect the specific organ system (eg, hoarseness in laryngeal amyloid, gross hematuria in genitourinary amyloid, and subconjunctival masses, often mistaken for lymphomas). The large majority of cases of localized amyloid that have been characterized biochemically appear to be AL, with evidence in some cases that the aggregates are being synthesized by clonal contiguous plasma cells. Even though patients are routinely evaluated for systemic involvement, disease may remain stable or only extend locally even with prolonged periods of observation, with cures reported if full resection is feasible.15
CUTANEOUS AMYLOIDOSES16
MACULAR AMYLOIDOSIS
MACULAR AMYLOIDOSIS
Macular amyloidosis commonly presents in the interscapular region as a pigmented patch of varying size. It can occur in other sites, such as the extensor surface of the arms, thighs, and shins. Typical is a rippled salt-and-pepper appearance with alternating hyperpigmentation and hypopigmentation (Fig. 125-6). It is more common in women and patients with a darker complexion. The pathology of macular amyloidosis is subtle and easily missed without clinical correlation.
EPIDEMIOLOGY COMMONLY AFFECTED SITES CLINICAL APPEARANCE PATHOLOGIC FEATURES
Macular amyloidosis ∼35% of cutaneous amyloidosis cases; women of darker complexion more commonly affected
Interscapular region, extensor surfaces of arms, thighs, and shins
Lichen amyloidosis ∼35% of cutaneous amyloidosis; most common subtype; men in their fifth to sixth decades are more commonly affected
Shins and forearms, occasionally upper back
∼15% of cases Characteristics of
∼15% of cases Characteristics of both macular and lichen amyloidosis
Biphasic
Biphasic cutaneous amyloidosis
cutaneous amyloidosis
both macular and lichen amyloidosis
2262
“Salt-and-pepper” appearance with alternating hyper- and hypopigmentation
Mild epidermal hyperkeratosis; collections of amyloid “corpuscles” within papillary dermis; melanincontaining histiocytes encircle the deposits
Linear rows of firm, pigmented, grouped, hyperkeratotic papules that can evolve into plaques
Epidermis is acanthotic and papillomatous with basal layer hyperpigmentation and elongated rete ridges; small collections of amphophilic material in papillary dermis surrounded by melanophages
Overlapping clinical
Overlapping pathologic features
Overlapping clinical features Overlapping pathologic features
features
Within the papillary dermis are small collections of amyloid globules, or “corpuscles.” These can be irregularly scattered and not found in every rete. A useful clue (as in lichen amyloidosis) is the presence of rare
21
melanin-containing histiocytes encircling these deposits. Often, special stains are necessary to demonstrate the amyloid. The extent of epidermal changes is minimal with only mild hyperkeratosis (Fig. 125-7).
LICHEN AMYLOIDOSIS
LICHEN AMYLOIDOSIS
Lichen amyloidosis is the most common type of cutaneous amyloidosis. It usually presents later in life, predominantly in the fifth and sixth decades, and is more common in men and patients with a higher Fitzpatrick skin type. The initial symptom of this disorder is intense pruritus that may improve with sun exposure and worsen during periods of stress. Hyperpigmented lesions are presumed to be secondary to scratching. Clinically, lesions often occur on the shins and forearms as linear rows of firm pigmented grouped hyperkeratotic papules that can evolve into large plaques (Fig. 125-8); the upper back also can be involved. As a result of the intense pruritus, the epidermis is often acanthotic and papillomatous with a compact horn; hyperkeratosis, hyperpigmentation of basal keratinocytes and elongation of rete ridges is typical. Basal cell vacuolar changes may occur with intraepidermal cytoid bodies. Within or adjacent to the lesion, changes of lichen simplex chronicus may occur. Within the broadened papillary dermis, there are small collections of amphophilic material often surrounded by melanophages (macrophages with engulfed melanin) (Figs. 125-9 and 125-10).
BIPHASIC CUTANEOUS AMYLOIDOSIS
BIPHASIC CUTANEOUS
AMYLOIDOSIS
Biphasic cutaneous amyloidosis refers to the concurrent presence of macular amyloidosis and lichen amyloidosis. This entity may also exist as a form in combination with blisters and poikilodermic skin lesions.19
2263
21
VARIANT FORMS OF LOCALIZED CUTANEOUS AMYLOIDOSIS
VARIANT FORMS OF
LOCALIZED CUTANEOUS
AMYLOIDOSIS
■ Towel-associated amyloidosis induced by prolonged contact with nylon towels.20
■ Anosacral amyloidosis has been described mainly in Chinese and Japanese patients presenting with
2264
brownish patches of plaques in the anosacral region. It is often associated with lichenification, and most patients report pruritus. Amyloid deposits are found in the papillary dermis with pigment incontinence.21
■ Notalgia paresthetica amyloidosis isolated sensory neuropathy that typically occurs on the back may be associated with amyloid, likely resulting from prolonged pruritus and scratching.22
■ Friction melanosis or amyloidosis prolonged contact with sponges, towels, plant sticks and leaves. In the opinion of the authors and others, these most likely represent variants of macular or lichen amyloidosis, with a similar pathology23,24
■ Lichen amyloidosis of the auricular concha hyperpigmentation of the bowl of the ear; likely a variant of lichen amyloidosis.25
NODULAR AMYLOIDOSIS
NODULAR AMYLOIDOSIS
Patients present with 1 or more deep-seated lesions that have a waxy or nodular appearance. They can appear as papules, nodules, or plaques (see Fig. 125-5). Common sites include the feet and trunk.26 Deposition may occur as discrete amorphous areas between collagen bundles. Nodular amyloidosis may be unique to the skin, although vascular involvement is common (Fig. 125-11). AL is the amyloid type of the large majority of cases that have been characterized biochemically, and the presence of monotypic plasma cells contiguous to nodules has suggested local synthesis (Fig. 125-12); nevertheless, followup for the development of a plasma cell dyscrasia or lymphoproliferative disease is indicated for these patients. In addition, up to 25% of nodular amyloidosis may be associated with primary Sjögren syndrome, a disease in which there may be an increased incidence of non-Hodgkin lymphoma of mucosa-associated lymphoid tissue.27
Cutaneous amyloidomas of β2-microglobulin have also rarely been described in patients with dialysis amyloidosis.28
AMYLOID ADJACENT TO TUMORS
AMYLOID ADJACENT
TO TUMORS
Although not evident clinically, it is not uncommon to find collections of amyloid adjacent to keratocytic tumors such as basal cell carcinoma, Bowen disease, squamous cell carcinoma, or associated with more benign lesions such as seborrheic or actinic keratoses. Between 66% and 77% of basal cell carcinomas, particularly the nodular type, have been reported to have amyloid deposits within the tumor stroma.29,30
21
CUTIS LAXA IN FAMILIAL AMYLOID CAUSED BY MUTANT GELSOLIN
CUTIS LAXA IN FAMILIAL
AMYLOID CAUSED BY
MUTANT GELSOLIN
Originally described in Finland as Meretoja syndrome, the gelsolin mutation has a worldwide distribution and manifests as lattice corneal dystrophy, Type 4, cranial and peripheral neuropathies, and, in some families, prominent renal involvement. Cutis laxa starts on the face, usually in the fifth decade of life, and is apparent as premature aging, but then becomes more generalized. The consequences of skin atrophy may be xerosis, easy bruising with minor trauma, loss of body and scalp hair and occasional lichen amyloidosis. Gelsolin amyloid tends to deposit adjacent to basement structures, including the subdermis and eccrine sweat glands, in dermal blood vessels, and between collagen and elastic fibers.9
TRANSTHYRETIN AMYLOIDOSIS
TRANSTHYRETIN
AMYLOIDOSIS
Amyloid can be found in the walls of small blood vessels, freely in the dermis as small fissured collections, and diffusely in adipose tissue between adipocytes (Fig. 125-13).8
INJECTION-SITE AMYLOIDOSES
INJECTION-SITE
AMYLOIDOSES
Localized forms of amyloid that result from therapeutics with amyloidogenic propensity have been found in association with insulin pumps and with the use of
2265
21
enfuvirtide (Fuzeon). Enfuvirtide is a second-line antiretroviral agent used in patients with multidrug resistance to HIV. It comes in a form that is reconstituted for subcutaneous injection, which may result in large, redyellow, subcutaneous nodules at the sites of injection. Biochemical characterization of these amyloids has been carried out by mass spectroscopy and sequence analysis, and fibrillogenesis demonstrated in vitro.31
AMYLOIDOSIS CUTIS DYSCHROMICA
AMYLOIDOSIS CUTIS
DYSCHROMICA
Amyloidosis cutis dyschromica is a rare condition in which patients develop progressive diffuse hyperpigmentation and hypopigmentation and atrophy (Fig. 125-14). It usually has no other associations, though occasional patients have neurologic changes, or signs and symptoms consistent with connective tissue disease. Pathogenesis is unknown, although photosensitivity has been implicated. Amyloid is usually localized within the papillary dermis, and stains with antikeratin antibodies.32,33
EPIDEMIOLOGY OF CUTANEOUS AMYLOIDOSES
Macular amyloidosis has a high incidence in Asia, the Middle East, and South America, but is rarely seen in Europe and North America. Macular lesions predominate in studies from Indonesia, Turkey, and India, although papular lesions were more common in one study from Kuala Lumpur. The highest incidence has been reported from southern China and Taiwan, and in most series, the female predominance is overwhelming. Familial cases are estimated to account for up to 10% overall, and. have been associated with specific
2266
gene defects, including mutations in the oncostatin M receptor (OSMR), interleukin-31 receptor A (IL31RA), and the RET protooncogene.17
Cutaneous lichen amyloidosis defines a variant of the multiple endocrine neoplasia 2A syndrome, which is primarily (∼95%) associated with medullary thyroid cancer (MTC), and less commonly with pheochromocytoma (30% to 50%) or primary hyperparathyroidism (20% to 30%) and mutations in the RET (rearranged during transfection) protooncogene, which causes constitutive activation of the transmembrane tyrosine kinase receptor. In this syndrome, lichen amyloidosis may occur in childhood before MTC, with most reported cases being the codon 634 mutations, which is estimated to give a 30% likelihood of developing lichen amyloidosis, with 77% affected individuals being women.34 In one Chinese family with MTC associated with cutaneous amyloidosis, disease was associated with a RET S891A mutation coexisting with a novel OSMR variant, G513D, in 3 family members, with clinical data suggesting a synergism between the 2 mutations that may be driving the clinical phenotype.35
GENETICS AND PRURITUS IN CUTANEOUS AMYLOIDOSIS
GENETICS AND PRURITUS
IN CUTANEOUS
AMYLOIDOSIS
Recent studies have identified receptors in skin additional to those responsible for histamine effects, including the Mas-related G-protein–coupled receptors, and the transient receptor potential vanilloid Type 1, some of which have been identified in sensory neurons and may respond to histamine, heat, or capsaicin.36
Particular interest has been directed to OSMR, which encodes OSMRb, a component of both the OSM Type II receptor and the IL-31 receptor. IL-31 has been implicated in pruritus associated with atopic dermatitis, prurigo nodularis and cutaneous T-cell lymphoma (Sézary syndrome).37 Thirteen heterozygous mutations in OSMRb have been described in association with familial primary cutaneous amyloidosis, most in the extracellular fibronectin III repeat domains central to receptor dimerization with gp-130 (IL-6) or IL-31RA. One heterozygous missense mutation in IL-31RA also has been reported in association with familial lichen amyloidosis.38 Lesional skin was found to have increased apoptosis of keratinocytes, epidermal hyperinnervation, cytokeratin-5 specific antibody reactivity, increased epidermal expression of IL-31 receptors, and elevated tissue/serum IL-31 levels.39,40
Although keratinocyte apoptosis appears to be a central event in the pathogenesis of lichen amyloidosis, and cytokeratin immunohistology a valuable adjunct to diagnosis, recent studies provide evidence that the amyloid fibrils in this disorder may be attributed to subsequences of galectin 7 (Gal7). Early extraction studies yielded codepositing keratin epitopes, as well as P-component, actin, and apoE (enriched in apoE4 in 2 studies), and Gal7, results which could
be confirmed by immunohistology17,41; however, only Gal7 was found to bind thioflavin T. This protein is one of a family of β-galactoside–binding proteins, only expressed in stratified epithelia, particularly abundant in the stratum spinosum, through which postmitotic keratinocytes move from the basal layer of the epithelium and transform into flattened, enucleated cells connected by tight junctions that secrete keratins to the extracellular matrix; it is markedly induced during keratinocyte apoptosis. Subsequences of Gal7 were found to be fibrillogenic in vitro, corresponding to fragments generated by enzymes known to be active during apoptosis, and modulated by coassociated actin.42 The exact relationship of these findings to keratinocyte apoptosis and the clinical symptoms of itch are active areas of investigation.
AN APPROACH TO THE DIAGNOSIS OF CUTANEOUS AMYLOIDOSIS
INITIAL EVALUATION
INITIAL EVALUATION
Patients with cutaneous amyloidosis may present to dermatologists for evaluation, or as referrals from specialists who suspect amyloid involving the skin. The specialist referral might be an individual in whom the
■History: evidence of systemic disease, and which organ systems may be involved; onset of disease in general and cutaneous lesions/pruritus in particular
■History: evidence of systemic disease, and which organ systems
may be involved; onset of disease in general and cutaneous lesions/pruritus in particular
■Family history: include ethnicity, and familial clustering of conditions known to be associated with amyloid
■Family history: include ethnicity, and familial clustering of condi-
tions known to be associated with amyloid
■Laboratory: immunoglobulin clonality for AL, elevated acute-phase reactants for AA, low TTR levels for ATTR
■Laboratory: immunoglobulin clonality for AL, elevated acute-phase
reactants for AA, low TTR levels for ATTR
■Genetic testing: TTR, MEFV, RET, others in specialty laboratory test results
■Genetic testing: TTR, MEFV, RET, others in specialty laboratory test
results
■Skin lesions: waxy deposits, macroglossia or lingual nodules; nodules, macules, lichen, and variants; single or multiple lesions; evidence of increased vascular fragility; hyperkeratosis/hyperpigmentation; cutis laxa
■Skin lesions: waxy deposits, macroglossia or lingual nodules;
nodules, macules, lichen, and variants; single or multiple lesions; evidence of increased vascular fragility; hyperkeratosis/hyperpigmentation; cutis laxa
■Biopsy: lesional skin or abdominal fat pad; Congo red or thioflavin T; subpapillary amyloid or congophilic angiography/involvement of adnexa
■Biopsy: lesional skin or abdominal fat pad; Congo red or thioflavin
T; subpapillary amyloid or congophilic angiography/involvement of adnexa
■Immunohistochemistry: generic (eg, P-component), fibril subunit specific (L-chain, TTR, GAL7), or coassociating (keratin, actin)-specific antibodies; immunoelectron microscopy
■Immunohistochemistry: generic (eg, P-component), fibril subunit
specific (L-chain, TTR, GAL7), or coassociating (keratin, actin)-specific antibodies; immunoelectron microscopy
■Mass spectroscopy: protein (AL variable; mutations) sequences and coassociating molecules
■Mass spectroscopy: protein (AL variable; mutations) sequences and
coassociating molecules
21
■Interscapular: lichen simplex chronicus; lichen sclerosis and atrophicus; postinflammatory hyperpigmentation; dorsal macular pigmentary incontinence; fixed drug eruption; notalgia paresthetica; poikiloderma; scleroderma/morphea
■Interscapular: lichen simplex chronicus; lichen sclerosis and atro-
phicus; postinflammatory hyperpigmentation; dorsal macular pigmentary incontinence; fixed drug eruption; notalgia paresthetica; poikiloderma; scleroderma/morphea
■Shins: prurigo nodularis; atopic eczema; pretibial myxedema; hypertrophic lichen planus
■Shins: prurigo nodularis; atopic eczema; pretibial myxedema;
hypertrophic lichen planus
■Nodules: cutaneous lymphomas; nevus lipomatosis; nodular cutaneous neoplasms; nodular pseudolymphomas
■Nodules: cutaneous lymphomas; nevus lipomatosis; nodular cuta-
neous neoplasms; nodular pseudolymphomas
■Waxy deposits: sebaceous keratosis; xanthomas
■Waxy deposits: sebaceous keratosis; xanthomas
■Periorbital purpura: dermatomyositis/heliotrope; posttraumatic raccoon eyes
■Periorbital purpura: dermatomyositis/heliotrope; posttraumatic
raccoon eyes
■Pinch purpura: vasculitic purpura; pigmented purpuric dermatoses; thrombocytopenic purpura; trichinosis
■Pinch purpura: vasculitic purpura; pigmented purpuric dermatoses;
thrombocytopenic purpura; trichinosis
diagnosis of systemic amyloid is suspected or known, and in whom skin lesions are to be examined and possibly studied pathologically; patients presenting to a dermatologist might include patients with cutaneous findings that include macular, lichen, or nodular amyloid in the differential diagnosis (Table 125-5). Initial evaluation should include a cataloging of comorbid medical conditions, such as carpal tunnel syndrome or other forms of neuropathy, enquiry regarding easy bruising, petechial rash, or ecchymoses, localized areas of pruritus on the upper back or shins, a search for multiple lesions, pinch or periorbital purpura, and an evaluation for macroglossia or lateral ridging of the tongue. Macular or papular lesions should be examined for a waxy appearance suggestive of amyloidosis. Some patients may be able to volunteer a family history of amyloid diseases; others may be aware of multiple relatives known to have neuropathy, renal or cardiac disease, or affected by periodic fever syndromes. A family history of cutaneous disease may be relevant in considering a patient with suspected macular or lichen amyloidosis from Southeast Asia or South America.
LABORATORY
LABORATORY
In addition to routine studies, specific laboratory testing may be suggestive for certain forms of systemic amyloidosis. For both systemic and localized AL amyloidosis, this includes a measurement of quantitative immunoglobulins (IgG, IgA, and IgM isotypes), serum and urine immunofixation, and a measurement of serum κ and λ free light chains, which will identify a clonal population of immunoglobulin light chains in more than 95% of patients with systemic AL disease, and a much lower number of patients with localized AL disease. For AA, an elevated erythrocyte sedimentation rate, as well as specific acute-phase reactants, such as C-reactive protein and fibrinogen, serve as surrogates for elevated levels of SAA protein, which can directly be measured in panels of acute-phase
2267
21
reactants available for profiling rheumatoid arthritis or inflammatory bowel disease. For suspected ATTR, an assay for serum TTR levels and TTR gene-sequence analysis may serve to identify a specific mutations, although it should be noted that up to 25% of patients also are found to have monoclonal immunoglobulins as an incidental finding.
PATHOLOGY
PATHOLOGY
Congo red binding to several forms of cutaneous amyloid may be weak and inferior to the use of thioflavin T or Pagoda red as diagnostic reagents; in addition, the interpretation of apple-green birefringence may be obscured by the small amount of amyloid in early lesions, and because of background white birefringence attributable to collagen in the dermis.30 To this end, a modified Congo Red immunohistochemical overlay has been suggested to improve diagnosis,43 and a luminescent conjugated oligothiophene, h-FTAA (heptamer formyl thiophene acetic acid), was developed to improve selectivity and sensitivity.44 The immunohistochemistry of amyloid in tissue has proven to be a valuable adjunct to confirming proper diagnosis and guiding treatment, although its applicability to cutaneous amyloid presents certain caveats not found in other organ systems. A panel of polyclonal or monoclonal antibodies for the identification and typing of amyloid in tissue includes anti–P-component, anti-apoE, anti–κ Ig light chain, and anti–λ Ig light chain, anti-TTR, anti-AA, anti-β2m, and generic or specific anti-keratin antibodies. In particular, the utility of antibodies to P-component as an immunohistologic surrogate marker for amyloid in tissue may be limited by the fact that this protein is an elastic fiber microfibrillar sheath-associated protein in normal skin,45 as well as in cutis laxa associated with Meretoja syndrome,9 and other dermal elastolytic conditions. Anti-TTR antibodies have been used to uncover amyloid in skin of patients with advanced ATTR (Fig. 125-15), and anti–keratin antibodies to type and define the distribution of localized cutaneous amyloidosis (Fig. 125-16). Among the organ-limited cutaneous amyloidoses, association with basal keratinocytes has been reflected in the identification of cytokeratin (CK)-5 as a major constituent of amyloid deposits and suggested a role of keratinocyte apoptosis in pathogenesis. In addition, other cytokeratins are positive immunochemically to varying degrees in lichen amyloidosis (CK5 > 1 > 14 > 10) and cutaneous amyloidosis associated with tumors and keratoses (CKL5 > 1 > 10 > 14).17 For all forms of localized cutaneous amyloid, diagnosis may be improved by immunohistochemistry using pan CK antibodies, or monoclonal reagents, such as 34βE12 (anti–keratin 903), which is reactive with CK5, CK1, CK10, and CK14.46 The identification of deposits as fibrils distinct from collagen or elastic fibers can be established by electron microscopy (Fig. 125-17). The utility of anti-κ or anti-λ antisera is limited by their specificity for determinants in the constant region of the Ig light chain, which may be lost during the proteolysis that is a concomitant of fibril
2268
formation; improved specificity has been reported by immunofluorescence, using specific imaging dyes, and by immunoelectron microscopy. Treatment of tissue by autoclaving with potassium permanganate causes loss of Congo red affinity for AA and Aβ2m; prolonged autoclaving affects binding of AL but not ATTR.47 Although immunohistochemistry has been validated as a technique of typing amyloids in tissue section, antibodies specific for some forms are not yet commercially available, and the importance of in-house quality controls has been emphasized.43
BIOCHEMISTRY
BIOCHEMISTRY
The diagnostic value of skin biopsy as a noninvasive way to diagnose systemic amyloids was first reported in the 1970s, and has been perfected as the abdominal fat pad aspiration or fat pad biopsy, both of which are office procedures with minimal morbidity.48 Aspiration has been used to screen for Congo red–positive material among fat globules (so-called fat rings), and can be modified for proteomic studies, quantitation of specific precursor proteins, and immunoelectron microscopy49; biopsy can be used to assess the morphology of amyloid in subcutaneous fat, for immunohistochemistry, and to isolate and sequence amyloid proteins by mass spectroscopy.50 Using this technique, the diagnosis of AL can be established in 80% to 90% of cases, and AA in 75% to 80%, with a lower (∼40%) yield for ATTR.49
Laser capture microdissection of amyloid from formalin-fixed amyloidotic tissue, followed by trypsin digestion, mass spectroscopy, and direct sequence analysis of peptides, has been validated for the typing of amyloid in various tissues, including cutaneous lesions and abdominal fat pad biopsies. This methodology also has been adapted for the identification of pathogenic mutations in cases of hereditary amyloid and for the identification of Ig light chain subgroups in systemic and localized AL amyloidosis.51 It also provides a profile of colocalizing proteins, such as P-component, keratins, and apoE, which may be important in pathogenesis.
TREATMENT STRATEGIES FOR AMYLOID DISEASES
SYSTEMIC AMYLOIDS
SYSTEMIC AMYLOIDS
The onset of amyloid deposition may be preceded over many years by the presence of a mutant precursor protein, excess Ig light chain, or elevated SAA levels in blood; functional abnormalities, such as pruritus, may also anticipate the appearance of characteristic skin lesions or MTC in familial cases of lichen amyloidosis. Identification of genetic abnormalities, either reflecting fibril subunit proteins, proteolysis, or coassociating conditions, allows for the potential to identify persons at risk and initiate prophylactic therapies early in the course of disease. For the systemic amyloidoses, lowering
21
the level of precursor protein correlates with clinical improvement and even regression of amyloid for SAA (with anticytokine therapy) and AL (with chemotherapies largely adapted from evolving myeloma protocols); in particular, the use of anti–IL-6 for AA amyloidosis, and high-dose dexamethasone, melphalan, thalidomide analogs, proteasome inhibitors, and anti-CD38 monoclonal antibodies have proven effective in reducing Ig light chain production by aberrant plasmablasts in AL amyloid.13,52 Ongoing trials have demonstrated the ability of silencing RNAs and antisense oligonucleotides to knock down the levels of mutant and wildtype TTR in blood, and are assessing efficacy for the clinical manifestations of polyneuropathy and/or cardiomyopathy. An alternative approach that has reached licensing is to use TTR tetramer stabilizers (diflunisal/tafamidis) to prevent dissociation to the monomer stage, which is the direct precursor of oligomers, amorphous aggregates, and fibrils in ATTR.53
An alternative strategy is to focus on inhibiting the aggregation phenomena responsible for oligomerization or fibril formation. This may occur at the deposition phase of amyloid formation, or to interfere with formed fibrils by intercalating at the sites of intermolecular contact critical for β-pleated sheet formation. Agents that may be active as fibril disrupters that have progressed to clinical testing include (a) doxycycline (ATTR, AL, Aβ2m), which disrupts fibril formation and mature fibrils, as well as inhibiting matrix metalloproteinase (MMP)-9; and the nutraceuticals54; (b) epigallocatechin- 3-gallate (green tea) (ATTR, Aβ-Alzheimer disease; AApoA2, transforming growth factor (TGF) β1-lattice corneal dystrophy, Type 1), which disrupts mature fibrils and suppresses markers of oxidative stress; and (c) curcumin (ATTR; Aβ), which induces oligomerization to a nontoxic “off pathway.”55 Low-molecularweight aggregation inhibitors include β-sheet breaker peptides, Fab, scFv or single chain camelid nanobodies, and peptide inhibitors of adhesive segments.56
A third approach is to use immunotherapy to neutralize oligomers and facilitate the clearance of tissue amyloid. Critical to this approach is to design antibodies that are specific for shared conformational epitopes rather than the native precursor protein, accessibility to the target conformers, and the safe elicitation of macrophagedependent clearance mechanisms. Currently, an anti-AL antibody with specificity for amyloid and oligomers is undergoing Phase III clinical trials, and antibodies to P-component are being tested as generic agents for systemic amyloids following depletion of serum, but not tissue amyloid–associated, SAP with a proline-derived small molecule that binds the pentamers together and facilitates their clearance from blood.57,58
THERAPY FOR CUTANEOUS AMYLOIDOSES
THERAPY FOR CUTANEOUS
AMYLOIDOSES
Currently, treatment of cutaneous manifestations associated with systemic amyloids has followed the
2269
21
general strategies outlined above.17 However, accessibility of skin amyloid allows for direct delivery to the sites of pathology. Early studies used dimethylsulfoxide (DMSO), based on a suggestion that it might be able to disaggregate AL fibrils, possible antiinflammatory effects, and deep permeability at the sites of application; general applicability, however was limited by the acrid smell and difficulty in obtaining preparations that were free of contaminants. Newer studies in patients with macular and lichen amyloidosis have reported rapid and significant improvement in pruritus and hyperpigmentation, although not resolution of amyloid on followup biopsies.59
The main concern for nodular amyloid is the potential for progression to systemic AL, which has been reported as up to 50%; therefore, the main therapeutic challenge is to assess for monoclonal gammopathy and for other organ involvement. More rarely, evaluation of nodular amyloidosis may reveal that a patient has primary Sjögrens disease, a syndrome characterized by dry eyes and dry mouth (sicca syndrome) sometimes associated with major hematologic, pulmonary, GI or renal involvement. This may in turn suggest the use of systemic treatments for sicca (eg, sialogogues) or Disease Modifying Agents (DMARDs) such as hydroxychloroquine or methotrexate for systemic disease. Absent any indication of systemic AL, treatment is determined by the size, location, and multiplicity of the cutaneous nodules. Lesions in locations such as the foot or temple have been effectively treated with curettage, excision, and, rarely, intralesional methotrexate or ablative laser therapy; a 9% risk of local reoccurrence has been reported, and long-term followup recommended if localized AL is documented by special studies.17,26
Treatment of localized cutaneous amyloidoses overlaps the spectrum of macular and papular (lichen) variants, with a specific target of interrupting the cycle of pruritus, scratching, and lichenification. Common treatments that have been used in small series include topical and intralesional steroids, topical calcineurin inhibitors, low-dose oral cyclophosphamide, dermabrasion, and cyclosporine. Small series have reported successes with carbon dioxide resurfacing surgical laser, yttrium-aluminum-garnet laser, tocoretinate (a synthetic esterified compound of tocopherol and retinoic acid), narrowband ultraviolet B light therapy, and a combination of psoralen and ultraviolet A with acitretin. Resurfacing, neodymium-doped yttrium-aluminum-garnet, and pulsed-dye laser treatments have been recommended because of the lack of scarring or pigmentary changes; in some instances, improvement in histology and resolution of amyloid have been reported.60
In general, antihistamines are ineffective for the treatment of pruritus in this disorder, likely reflecting the lack of generation of histamine or involvement of histamine receptors in observed pathology. An alternative focus has been on the common occurrence of dysesthesias correlating with lichenoid skin lesions, and changes in epidermal nerve fiber density that have been demonstrated on biopsies. Topical capsaicin
2270
0.025%, which releases neuropeptides from C-fibers, and prevents their deposition, may be tried, and there may be role for inhibitors of nonhistamine pruritogens and receptors currently being considered.36 Lastly, antagonists of IL-31 receptors or its receptor have been under development for the treatment of atopic dermatitis, and might also have relevance to ameliorating the itch of lichen amyloidosis.37
ACKNOWLEDGMENTS
Research support from the Seaver Foundation is gratefully acknowledged.
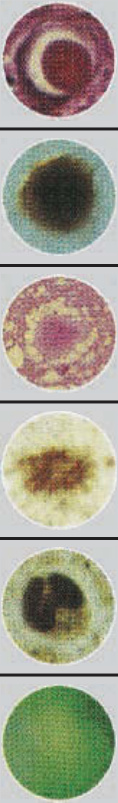
Figure 125-1 Alternative conformations of amyloidogenic proteins, including amorphous aggregates, oligomers and fibrils, and factors implicated in fibrillogenesis. Amyloidosis within the spectrum of aggregation disorders, several of which are implicated in neurodegeneration. (Adapted by permission from Springer Nature: Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16(6):574-81. Copyright © 2009.)
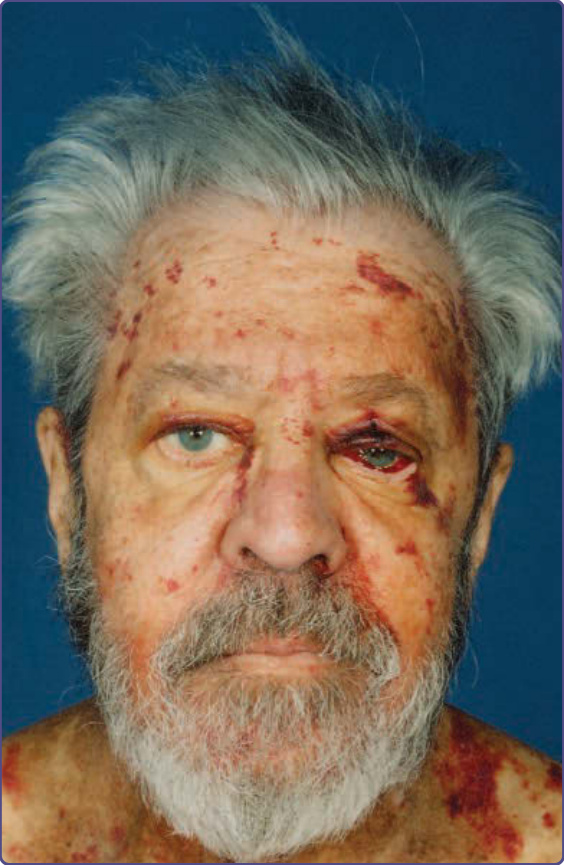
Figure 125-2 Pinch, periorbital and facial purpura in a patient with known systemic AL amyloidosis. Patient grew a beard to avoid the trauma/purpura associated with shaving.
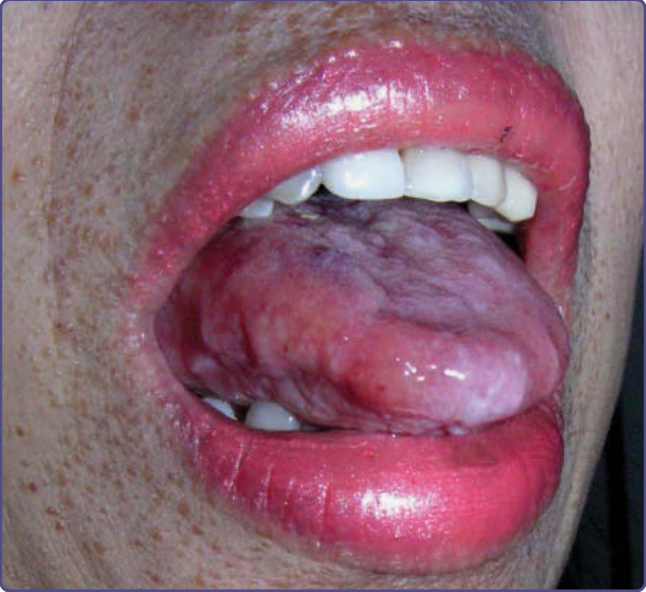
Figure 125-3 Macroglossia with lateral ridging caused by the impression of adjacent teeth in a patient with known systemic AL amyloidosis. Note the perioral macules with a waxy appearance also caused by cutaneous amyloid.
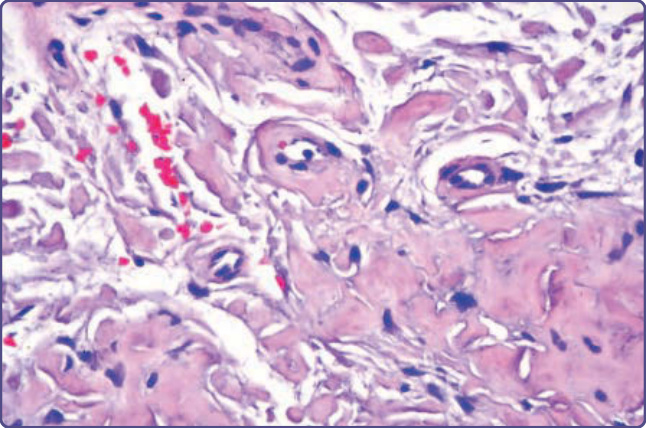
Figure 125-4 AL amyloidosis showing deposits in the dermis and walls of capillaries (hematoxylin and eosin, ×10 magnification).
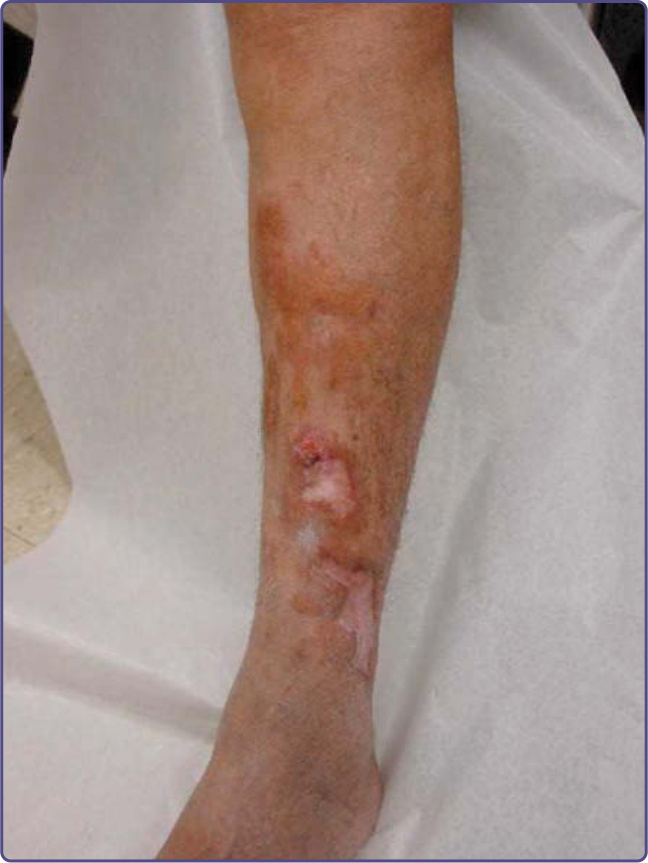
Figure 125-5 Multiple large and confluent nodules developed on the shin of a patient with nodular amyloidosis, noting the site of biopsy shown in Fig. 125-4. Followed over several years, the patient had no indication of systemic AL amyloid or plasma cell dyscrasia on repeated evaluations.
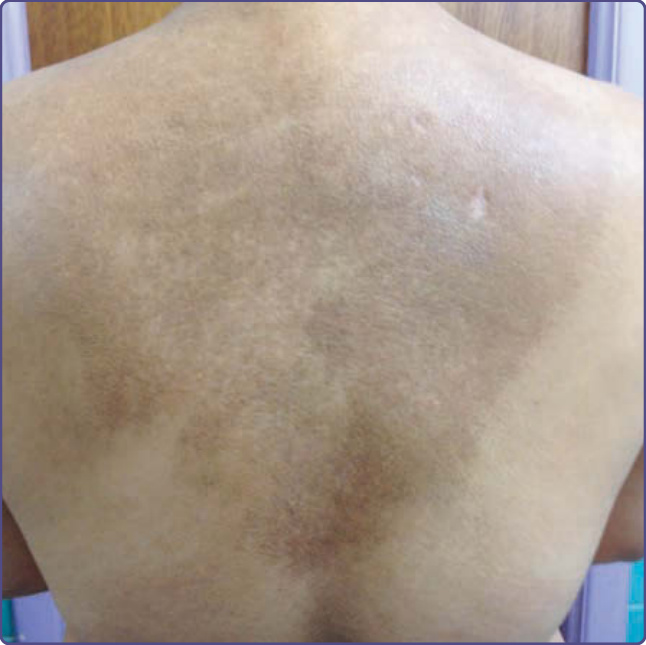
Figure 125-6 Interscapular area of a patient showing edge-shaped rippled hypopigmentation and hyperpigmentation consistent with macular amyloidosis.
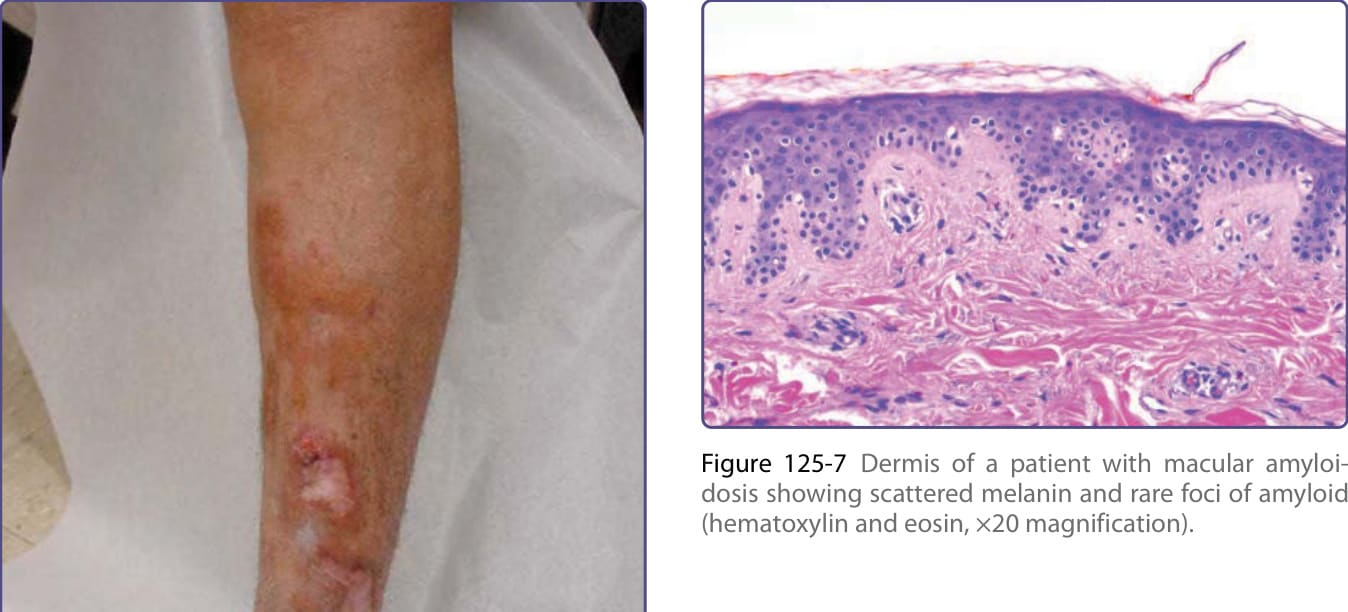
Figure 125-7 Dermis of a patient with macular amyloidosis showing scattered melanin and rare foci of amyloid (hematoxylin and eosin, ×20 magnification).
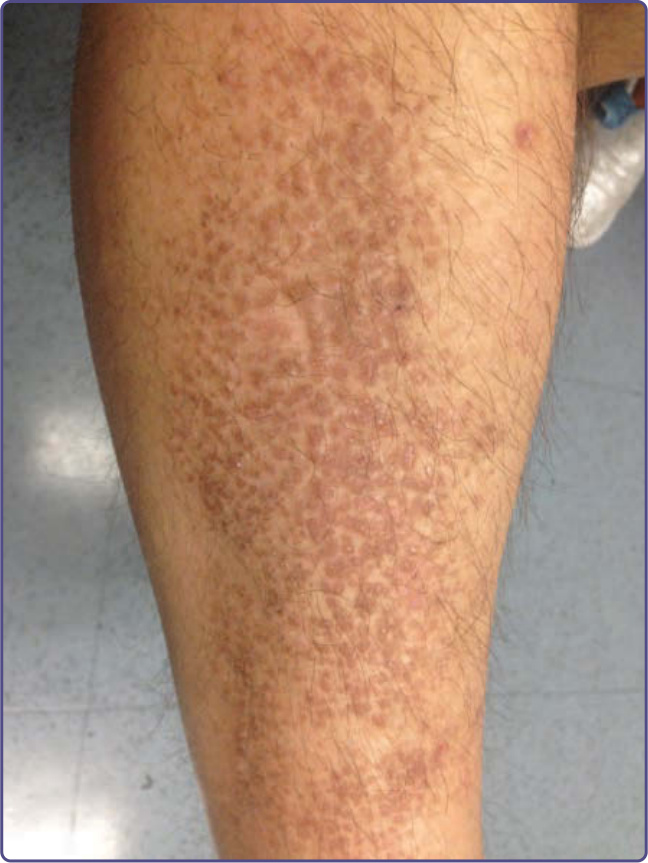
Figure 125-8 Linear keratotic papules on the shin characteristic for lichen amyloidosis.
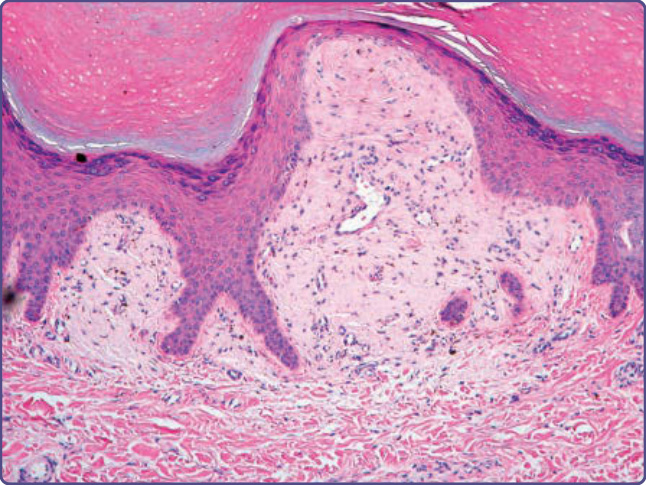
Figure 125-9 Beneath an acanthotic and papillomatous epidermis, a widened dermal papillae contains amyloid globules (hematoxylin and eosin, ×4 magnification).
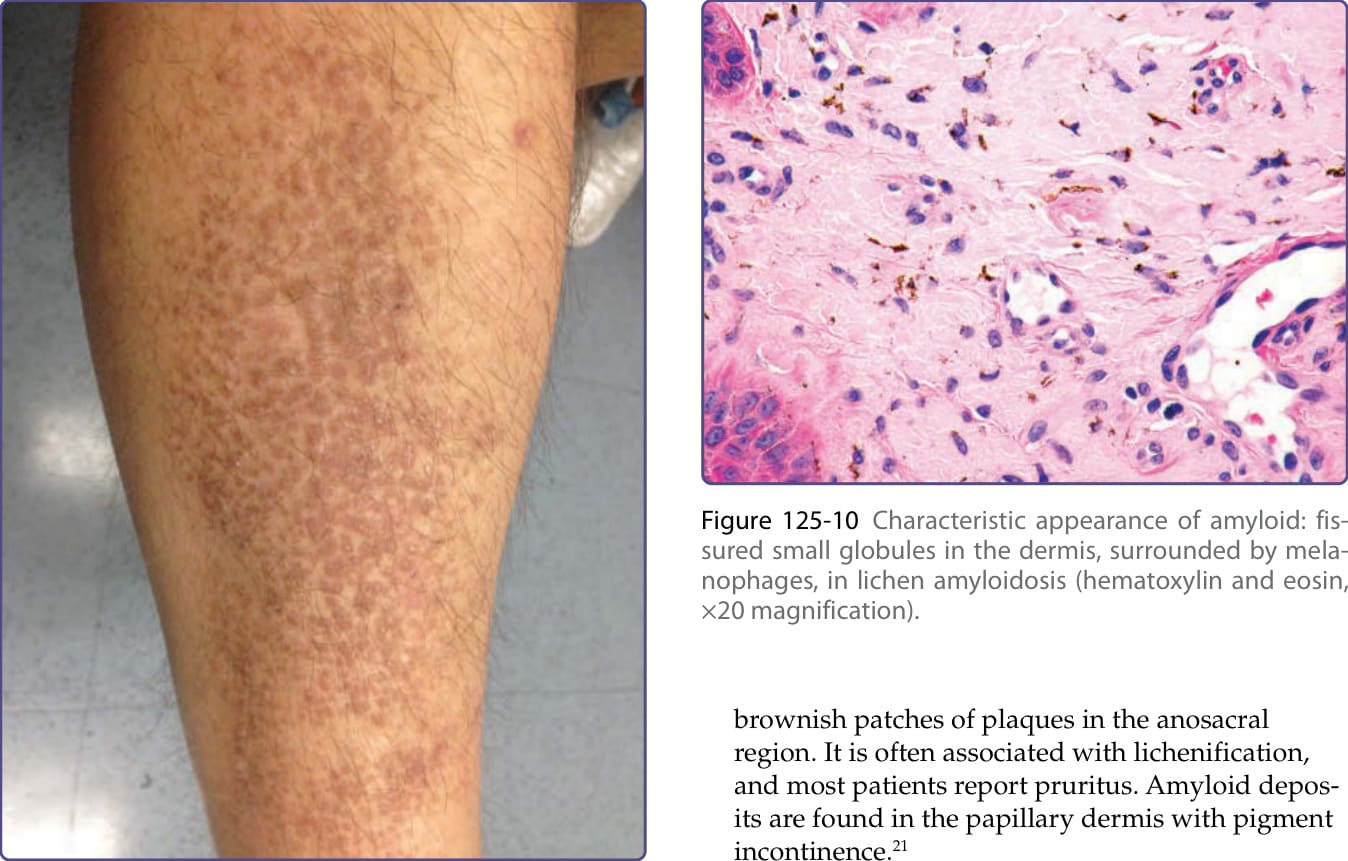
Figure 125-10 Characteristic appearance of amyloid: fissured small globules in the dermis, surrounded by melanophages, in lichen amyloidosis (hematoxylin and eosin, ×20 magnification).
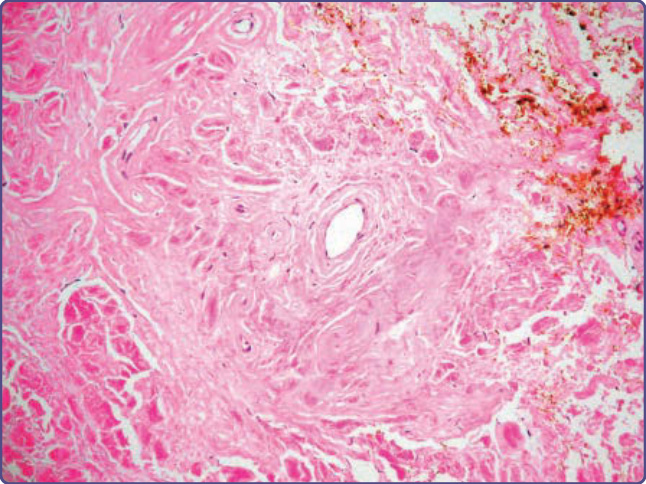
Figure 125-11 Diffuse deposits of amyloid completely replacing the dermis and surrounding blood vessels in nodular amyloidosis (hematoxylin and eosin, ×4 magnification).
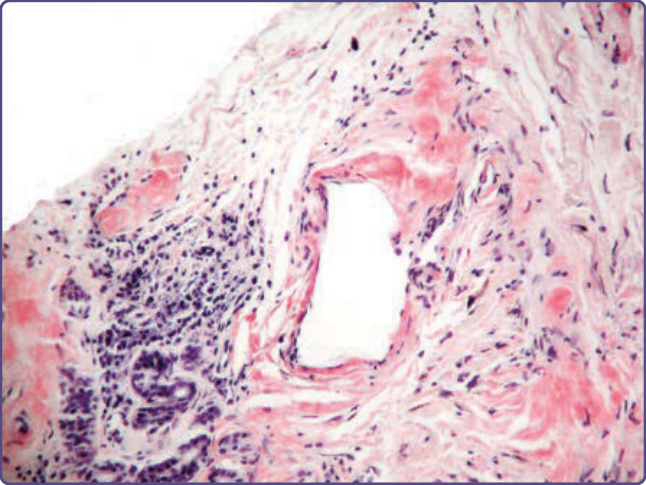
Figure 125-12 Small collection of plasma cells adjacent to nodular amyloid (Congo red, ×10 magnification). Immunohistology may reveal the same light chain in the amyloid as is produced by the adjacent plasma cells.
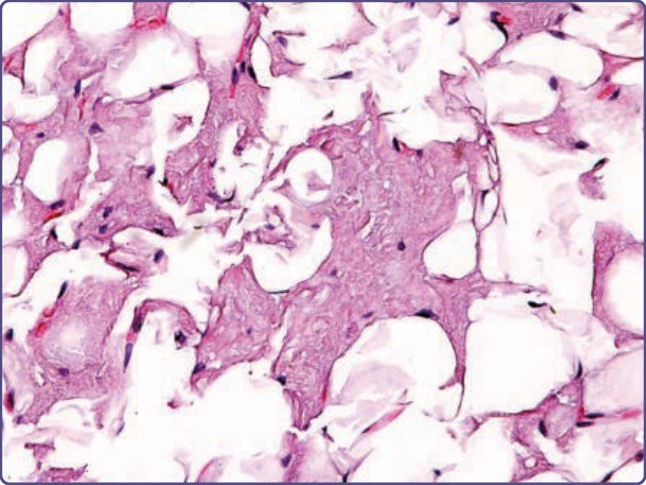
Figure 125-13 Transthyretin amyloid, late stage, apparent on abdominal fat pad biopsy, with fissured masses of amyloid replacing and surrounding adipocytes (hematoxylin and eosin, ×4 magnification).
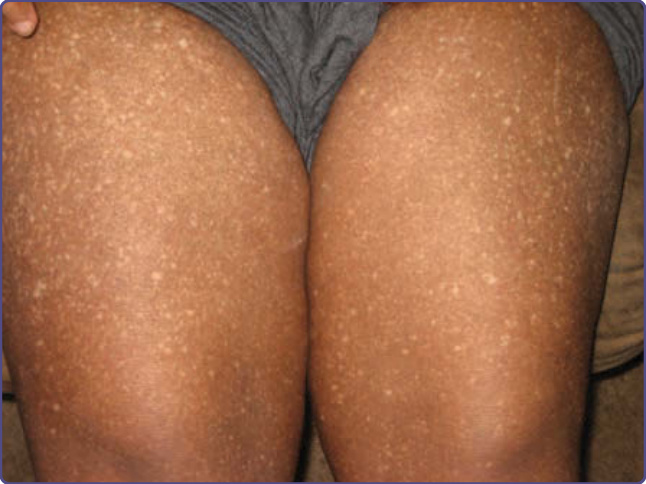
Figure 125-14 Spotted hypopigmentation on a hyperpigmented background in a patient with amyloidosis cutis dyschromica.
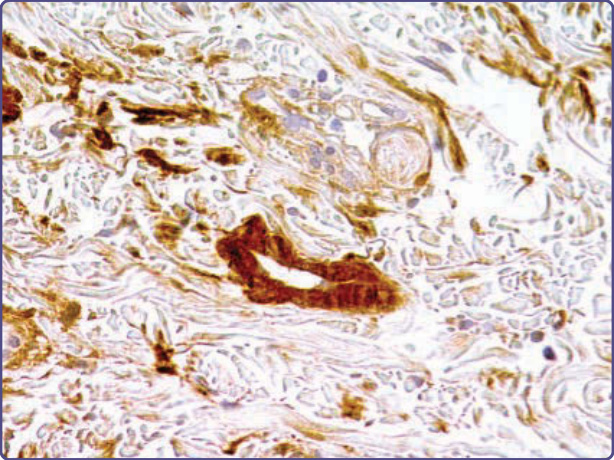
Figure 125-15 Anti-transthyretin (TTR) (prealbumin) antibodies highlighting a blood vessel in a patient with amyloidosis transthyretin (ATTR) amyloid attributable to familial amyloid polyneuropathy (hematoxylin and eosin (H&E), diaminobenzidine, ×10 magnification).
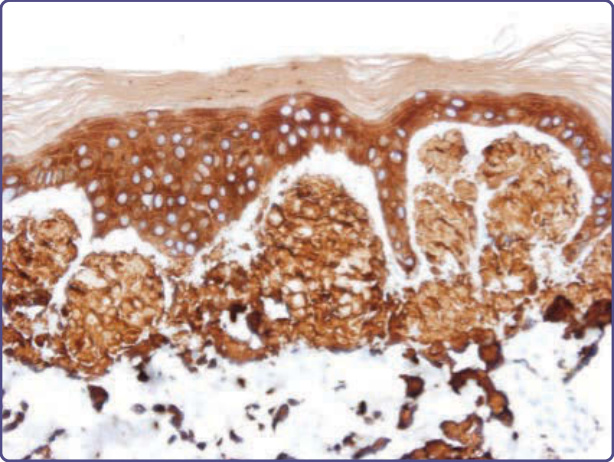
Figure 125-16 Anti–keratin 903 antibodies highlighting amyloid in expanded dermal papillae (hematoxylin and eosin (H&E), diaminobenzidine, ×4 magnification).
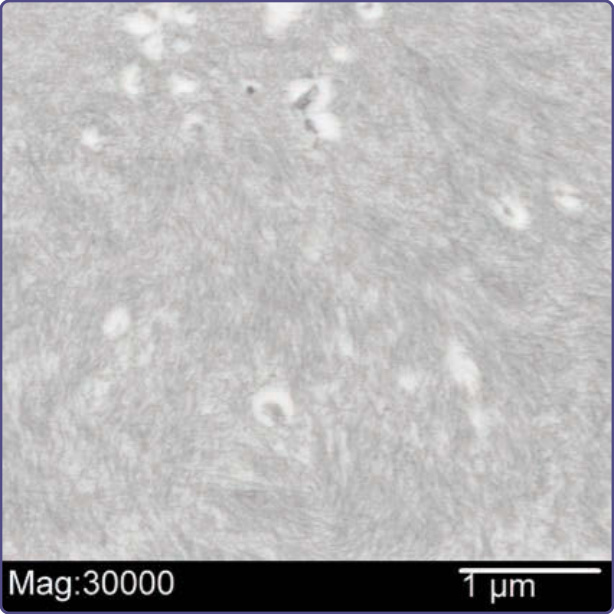
Figure 125-17 Electron micrograph showing thin filaments of amyloid in the dermis measuring 7 to 10 nm in thickness (uranyl acetate, lead citrate, ×30,000 magnification).

TABLE 125-1 Histologic Criteria for the Definition of Amyloid
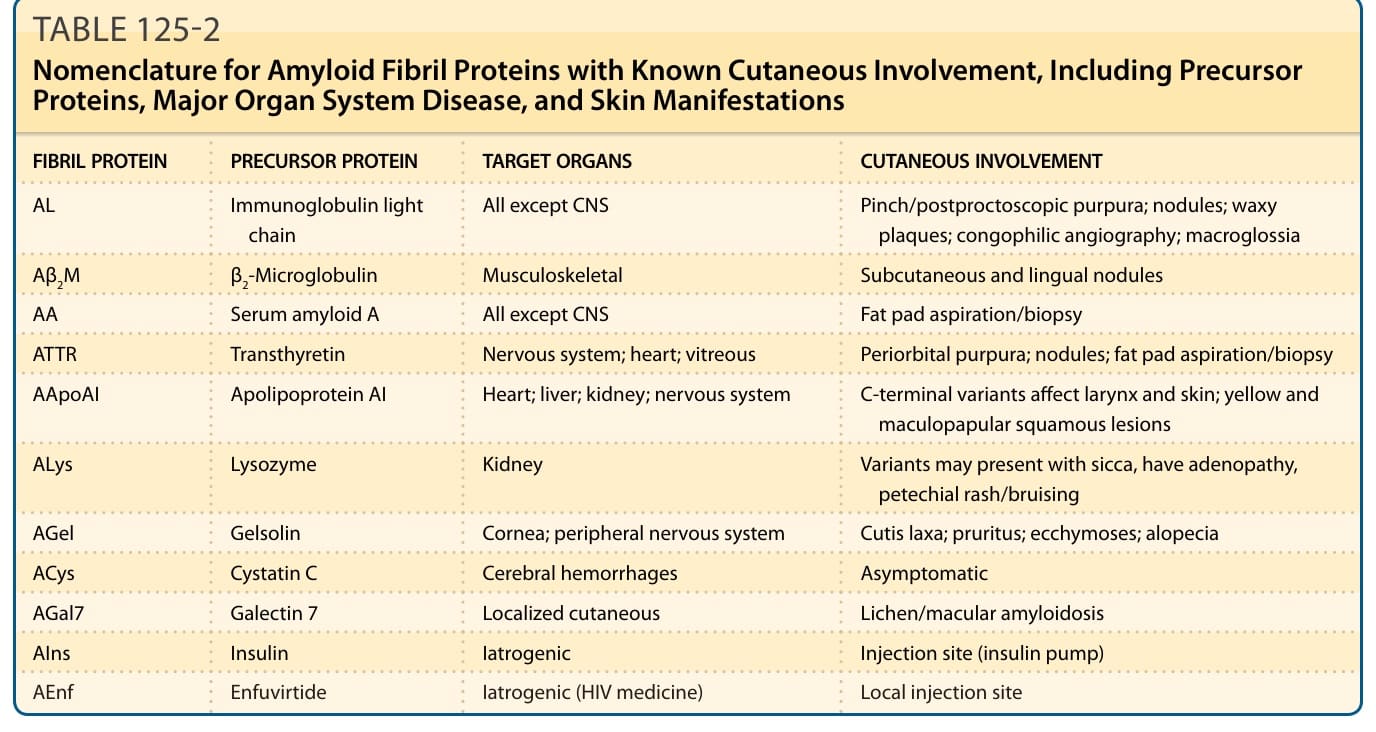
TABLE 125-2 Nomenclature for Amyloid Fibril Proteins with Known Cutaneous Involvement, Including Precursor Proteins, Major Organ System Disease, and Skin Manifestations
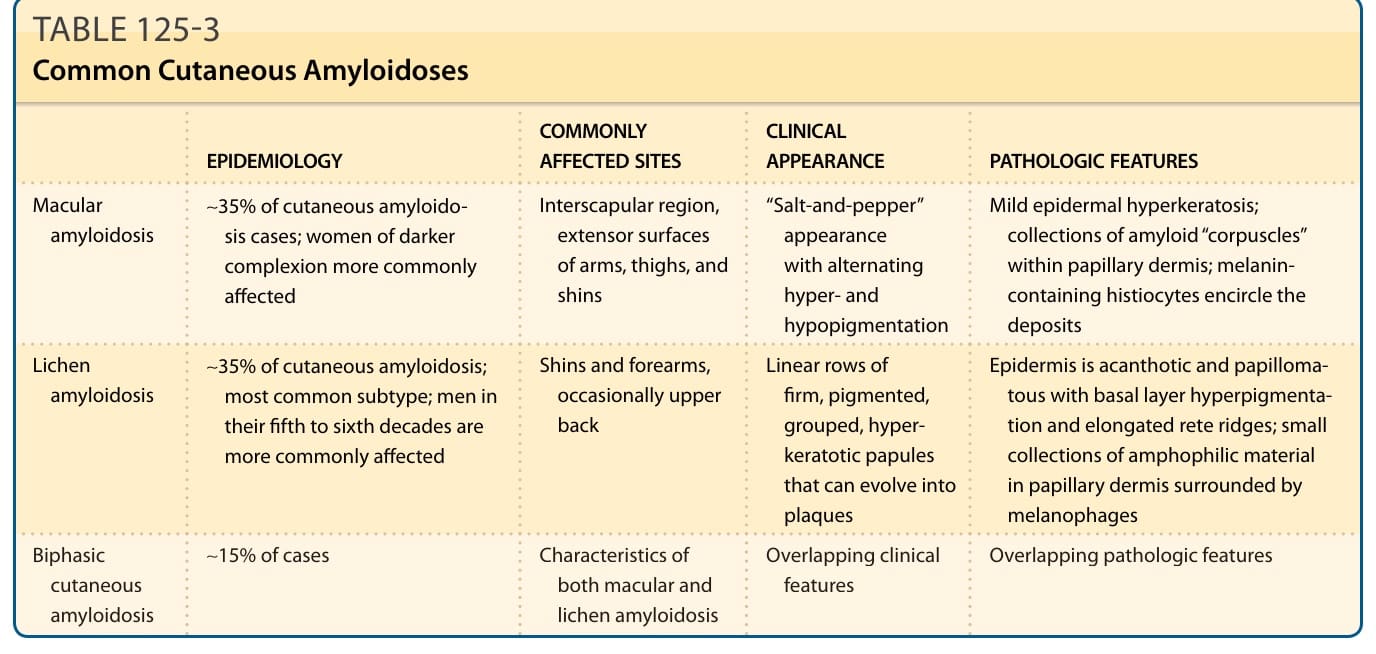
TABLE 125-3 Common Cutaneous Amyloidoses

Table 125-4 outlines the diagnosis of patients with known or suspected cutaneous amyloidosis.

TABLE 125-5 Differential Diagnosis of Cutaneous Amyloidoses