法布瑞氏病 (Fabry Disease)
PART 21
代謝性、遺傳性與系統性疾病 (Metabolic, Genetic, and Systemic Diseases)
重點一覽 (AT-A-GLANCE)
■ 在所有族群中,發生率估計介於 1:3200 至 1:170,000 人口之間。
■ 為一種 X 染色體連鎖 (X-linked) 的溶體儲積症 (lysosomal storage disorder)。
■ 在男性中具高度外顯率 (highly penetrant);女性異型合子 (heterozygotes) 的表現度 (expressivity) 變異很大。
■ α-半乳糖苷酶 A (α-galactosidase A) 部分或完全缺乏,伴隨醣鞘脂質 (glycosphingolipids,主要為三己醣神經醯胺 globotriaosylceramide) 的沉積。
■ 典型變異型主要影響皮膚、腎臟、心臟、眼睛與腦部。
■ 男性的預期壽命縮短 20 年,女性縮短 15 年。
■ 晚發型 (later onset) 變異型較輕微,且主要侵犯單一器官(例如腎臟或心臟)。
■ 皮膚科表現包括血管角化瘤 (angiokeratoma)、毛細血管擴張 (telangiectases)、「假性肢端肥大症」(“pseudoacromegalic”) 面容、少汗症 (hypohidrosis) 與多汗症 (hyperhidrosis)、淋巴水腫 (lymphoedema),以及雷諾現象 (Raynaud phenomenon)。
■ 光學顯微鏡 (light microscopy) 顯示真皮上層血管擴張 (ectatic upper dermal vessels)、邊緣表皮棘層增厚 (peripheral epidermal acanthosis) 與程度不等的過度角化 (hyperkeratosis)。
■ 電子顯微鏡 (electron microscopy) 顯示細胞質內、電子緻密、空泡狀的「斑馬小體」(“Zebra bodies”)。
■ 治療為症狀治療、酵素替代療法 (enzyme replacement);以及伴護蛋白療法 (chaperone therapy,用於可受惠之突變)。
流行病學 (EPIDEMIOLOGY)
法布瑞氏病(α-galactosidase A deficiency)一般被認為是僅次於高雪氏病 (Gaucher disease) 之後,盛行率第二高的溶體儲積症,¹,² 估計發生率介於 1:40,000 至 1:170,000 人之間。所有族群皆會受影響。新生兒篩檢研究顯示,GLA 基因突變的發生率更高,且部分受影響的個體罹患較輕微的疾病變異型,這些變異型與顯著的殘餘酵素活性相關。這類變異型可能以高得多的頻率出現(例如 1:3200),如近期一項北義大利新生兒調查所示。³ 一項針對日本超過 7000 名新生兒的研究指出,致病性突變的發生率約為 1:7000,⁴ 歐洲也報告了類似的發生率。⁵
這些變異型常以單一器官(例如心臟或腎臟)受侵犯為主,且於成年期發病。一項針對台灣華人族群的研究顯示,在新生兒中,心臟變異型法布瑞氏致病突變 IVS4+919G>A 具有出乎意料的高盛行率(男性約 1:1600),於特發性肥厚性心肌病 (idiopathic hypertrophic cardiomyopathy) 病人中亦然。⁶
病因與發病機轉 (ETIOLOGY AND PATHOGENESIS)
法布瑞氏病(線上人類孟德爾遺傳 Online Mendelian Inheritance in Man #301500)是一種罕見的 X 染色體連鎖代謝疾病,肇因於溶體酵素 α-半乳糖苷酶 A (α-galactosidase A) 的部分或完全缺乏(圖 127-1)。由於此酵素缺乏,帶有末端 α-半乳糖基殘基的中性鞘脂質(主要為三己醣神經醯胺 globotriaosylceramide [Gb3])會堆積於不同組織與體液的溶體中(包括腎臟腎小球與腎小管的上皮細胞;心肌細胞;自主神經系統的神經節細胞;角膜;血管的內皮細胞、周皮細胞 (perithelial) 與平滑肌細胞;以及結締組織的組織球與網狀細胞)。三己醣鞘氨醇 (globotriaosylsphingosine,為 Gb3 的去醯化衍生物) 濃度升高,與男女兩性的疾病嚴重度相關。三己醣鞘氨醇會抑制 α-galactosidase A 的活性,並有人提出女性體內濃度升高可能具有致病意義。⁷ 臨床發病時間變異很大(圖 127-2)。儘管本病為 X 染色體連鎖,異型合子女性 (heterozygous females) 仍經常受影響,且其嚴重程度可能與半合子男性 (hemizygous males) 相當。⁸ 女性的臨床症狀通常較男性晚約十年出現,且女性的器官損傷通常較男性輕微。女性的表現變異性可歸因於萊昂假說 (Lyon hypothesis),即女性體內一條 X 染色體會隨機被去活化 (inactivated),而另一條提供遺傳訊息。在有症狀的法布瑞氏病女性中,突變的 X 染色體較有可能在一系列組織中被表現。⁹
α-galactosidase A 的基因 GLA 位於 Xq22.1 區域。GLA 基因中已鑑定出超過 600 種突變,¹⁰⁻¹² 包括誤義 (missense) 突變、無義 (nonsense) 突變,以及單一胺基酸的缺失與插入。這些突變多為「私有」(“private”) 突變,僅在個別家族中被鑑定出。某些突變(例如 p.N215S 突變)與單一器官或晚發型變異相關,且這些病人儘管有法布瑞氏病的臨床證據,其尿中 Gb3 濃度卻正常。
三己醣神經醯胺的代謝路徑 (Metabolic pathway of globotriaosylceramide):α Gal A 將三己醣神經醯胺 (Globotriaosylceramide, Gb3)(結構為 Gal-Gal-Glu-Cer,含 α、β、β 鍵結)代謝為半乳糖 (Gal) 與乳醣神經醯胺 (Lactosylceramide,結構為 Gal-Glu-Cer,含 β、β 鍵結)。
皮膚表現 (CUTANEOUS MANIFESTATIONS)
皮膚血管病灶 (CUTANEOUS VASCULAR LESIONS)
血管角化瘤 (ANGIOKERATOMA)
血管角化瘤是法布瑞氏病的皮膚標誌性表現。¹¹⁻¹³ 血管角化瘤見於 70% 的男性與 39% 的女性,¹³,¹⁴ 為針尖大小至 4 mm 直徑、暗紅色至藍黑色的斑疹與丘疹病灶,按壓不會褪色 (do not blanch on pressure)(圖 127-3)。其上方的過度角化 (hyperkeratosis) 程度不一,在生殖器與肚臍以外的部位常缺如。
法布瑞氏病的自然病程 (Natural history of Fabry disease):依年齡演進依序出現心臟疾病 (Cardiac disease)、中樞神經系統疾病 (CNS disease)、腎臟疾病 (Renal disease)、胃腸道疾病 (GI disease);初始症狀 (Initial symptom) 包括角膜混濁 (Corneal opacities)、血管角化瘤 (Angiokeratomas) 與肢端感覺異常 (Acroparesthesias),多於 0 至 20 歲間出現,並延續至 40+ 歲。診斷時間 (Diagnosis):典型法布瑞氏病的平均診斷年齡為 50 歲(年齡範圍:40–56 歲),並影響生活品質 (Quality of life)。
血管角化瘤在男性中出現於 5 至 15 歲之間,在女性中出現於 8 至 25 歲之間。¹⁵⁻¹⁷ 它們是 352 名小兒病人中 50 名(14.2%)的表現特徵,¹⁸ 是 359 名成年男性中 114 名(31.7%)(n = 359)以及 118 名成年女性中 13 名(11.2%)(n = 118)的表現特徵。¹⁹ 病灶可為廣泛分布或群聚。在男性中,病灶典型位於「泳褲」(“bathing trunk”) 區域(生殖器、臀部、下腹部、肚臍、腹股溝、大腿內側與薦部)。血管角化瘤亦見於近端肢體(尤其是其內側面)、肘部與膝部、手掌與足底,以及手指遠端指節 (distal phalanges) 之上(圖 127-4)。病灶可能發生於嘴唇,尤其沿著唇紅緣 (vermilion border),偶爾發生於黏膜表面(圖 127-5)。它們很少出現於臉部其他位置。在女性中,血管角化瘤通常稀疏分布(圖 127-6),偶爾可能呈皮節 (dermatomal) 分布。¹³ 最常見的部位為軀幹與近端肢體。¹³ 女性生殖器病灶相對少見。
組織病理學 (HISTOPATHOLOGY)
組織學典型顯示真皮乳頭層 (papillary dermis) 內擴張、變寬的微血管 (dilated, ectatic capillaries),中央表皮程度不等地變薄,病灶邊緣處表皮棘層增厚 (epidermal acanthosis),其上方有程度不等的局部緻密正角化過度 (focal compact orthohyperkeratosis)(圖 127-7A)。內皮細胞、周皮細胞 (perithelial)、神經周圍細胞 (perineural)、外分泌汗腺 (eccrine)、平滑肌細胞與纖維母細胞 (fibroblasts) 內充滿了含醣鞘脂質的細胞質空泡,可用甲苯胺藍 (toluidine blue) 染色顯現。²⁰ 在電子顯微鏡下典型可見特徵性、電子緻密、層狀的細胞質內空泡狀內含物(斑馬小體 Zebra bodies)(圖 127-7B-D)。²¹ 它們呈現明暗交替、4 至 6 nm 條帶的型態。這些內含物可能出現於血管角化瘤或正常皮膚的切片中。它們在異型合子女性的皮膚中可能缺如。當其他診斷檢測結果不明確時,使用抗 Gb3 抗體 (anti-Gb3 antibodies) 的電子顯微鏡與免疫電子顯微鏡 (immunoelectron microscopy) 是有用的工具。²²
血管瘤與毛細血管擴張 (ANGIOMAS AND TELANGIECTASES)
多達三分之一的法布瑞氏病男性與三分之二的法布瑞氏病女性並無血管角化瘤。¹⁶,¹⁷ 其中一部分人完全沒有皮膚血管病灶,¹ 另一些人則有 1 至 2 mm 直徑、鮮紅色的斑狀血管瘤性病灶,臨床上可能代表早期血管角化瘤或斑狀血管瘤 (macular hemangiomas)。¹¹ 也有病人出現廣泛的丘疹狀血管瘤,臨床與組織學上與櫻桃狀血管瘤 (cherry angiomas) 一致。在一個系列中,以心臟表型為主(N215S 突變)的病人很少有典型血管角化瘤,但此組中三分之一的男性有廣泛(>100 個)的櫻桃狀血管瘤。²³ 由於櫻桃狀血管瘤在一般族群中的盛行率可能高達 50%,¹⁸ 其在法布瑞氏病病人中的意義並不明確。毛細血管擴張也很常見,¹³ 可藉由透壓鏡檢查 (diascopic pressure) 下會褪色 (blanching) 的特性,與血管角化瘤、斑狀血管瘤與櫻桃狀血管瘤區分。登錄資料顯示,毛細血管擴張在男性(23%)較女性(9%)更常見。¹³,¹⁴ 雖然是根據病人回憶,但資料顯示毛細血管擴張的出現晚於血管角化瘤,男性的平均發病年齡為 26 歲(SD 17,範圍:3–70 歲),女性為 42 歲(SD 22,範圍:5–73 歲)。¹³ 在多數病例中,毛細血管擴張發生於日曬部位,如臉部與頸部 V 形區,常見於膚質第 I 型或第 II 型的病人。偶爾,毛細血管擴張可見於不尋常的部位,如脅腹 (flanks)、腹股溝,以及肘部與膝部屈側。這些區域的皮膚鏡檢查 (dermoscopy) 顯示真皮上層血管擴張、迂曲 (dilated, tortuous upper dermal vessels)(見圖 127-3)。此情形傾向發生於有廣泛血管角化瘤且疾病完全表型表現的病人。
與疾病嚴重度的關係 (RELATIONSHIP TO DISEASE SEVERITY)
在兩性中,皮膚血管病灶(即毛細血管擴張與/或血管角化瘤)的存在,與較高的疾病嚴重度評分及較高的主要器官侵犯盛行率相關。¹³,¹⁴ 因此,在有皮膚血管病灶者中,腦血管侵犯(中風與短暫性腦缺血發作 transient ischemic attack)見於 38% 的女性與 32% 的男性,但在無皮膚血管病灶者中僅見於 12% 的女性與 9% 的男性。心臟侵犯在有皮膚血管病灶者中見於 80% 的女性與 73% 的男性,相對於無皮膚血管病灶者的 38% 女性與 49% 男性;腎臟侵犯在有皮膚血管病灶者中見於 62% 的女性與 72% 的男性,相對於無皮膚血管病灶者的 29% 女性與 42% 男性。在高血壓、眼、耳、胃腸道與其他神經學侵犯方面也觀察到類似的差異。¹⁴ 這些發現顯示了皮膚科評估的重要性,以及其在系統性罹病率方面可能的預測價值。
面部特徵 (FACIAL FEATURES)
某些法布瑞氏病家族曾被描述有「假性肢端肥大症」(“pseudoacromegalic”) 的面部外觀(圖 127-8)。此特徵已記錄於兩項較大型的研究中。一項由 3 位臨床遺傳學家組成的小組對 38 名病人進行的評估,係根據標準化醫學攝影 (standardized medical photography),²⁴ 並鑑定出(依出現頻率由高至低)眼周飽滿 (periorbital fullness)、突出的耳垂、濃眉 (bushy eyebrows)、後縮的前額、明顯的鼻角 (nasal angle)、寬大的鼻部/球狀鼻尖、突出的眶上脊 (supraorbital ridges)、淺平的中臉部、豐厚的嘴唇、突出的鼻樑、寬闊的鼻翼基部 (alar base)、粗獷的特徵、向後旋轉的耳朵,以及下顎前突 (prognathism)。第二項研究中,面部畸形以三維密集表面建模 (three-dimensional dense surface modeling) 與使用三維攝影測量相機 (three-dimensional photogrammetric camera) 的人體測量分析客觀評估,將 20 名男性與 22 名女性法布瑞氏病病人的面部特徵與對照組比較。²⁵ 此研究在男性中(女性中較不明顯)證實了上述面部特徵的存在。這些面部特徵的出現應促使進行適當的法布瑞氏病檢查。法布瑞氏病面容 (Fabry face) 在有廣泛血管角化瘤且屬嚴重端典型疾病的病人中似乎較常見。在 41 名典型疾病表型的男性中,26 名(63%)有中度或顯著的面部變化;這 26 名男性中有 16 名(61%)有瀰漫性軀體血管角化瘤 (angiokeratoma corporis diffusum)。相對地,在 19 名以心臟表型為主的男性中,僅 2 名(10.5%)出現極輕微程度的面部特徵(僅眼周浮腫 periorbital puffiness)。¹⁴
下肢水腫與淋巴水腫 (LOWER-LIMB EDEMA AND LYMPHEDEMA)
水腫與淋巴水腫,尤其影響肢體者,常見於法布瑞氏病病人(圖 127-9)。淋巴水腫在法布瑞氏病的原始描述中即被提及,並見於其他溶體儲積症,如 α-N-乙醯半乳糖胺酶缺乏症 (α-N-acetylgalactosaminidase deficiency)。²⁶ 病例報告記載其為本病不尋常的表現特徵,²⁷ 有些報告則指出家族性淋巴水腫 (familial lymphedema) 與法布瑞氏病可能有關聯。²⁸ 來自法布瑞結局調查 (Fabry Outcome Survey) 超過 700 名病人的登錄資料證實,可逆性下肢周邊水腫見於 25% 的法布瑞氏病男性與 17% 的女性;淋巴水腫見於 16% 的男性與 7% 的女性。平均發病年齡男性為 37 歲(範圍:13–70 歲),女性則約晚十年。¹³ 此比例顯著高於英國社區記載的盛行率(每 1000 人口 1.33 人)。²⁹ 在社區中,女性的發生率為男性的 5 倍。其變化背後的機轉並不清楚,且這些變化的存在與腎臟或心臟侵犯並不相關。可能的貢獻因素包括醣鞘脂質堆積、³⁰ 反覆水腫,以及淋巴管的原發性異常。³¹,³²
Amann-Vesti 等人使用螢光微淋巴管造影術 (fluorescence microlymphography) 與淋巴微血管壓力 (lymph capillary pressure) 測量技術,證明 5 名半合子男性中有 5 名、5 名異型合子女性中有 5 名出現微淋巴管網絡的破碎化 (fragmentation),而 12 名健康對照中則無一出現。即使在淋巴水腫尚未顯現時,皮膚初始淋巴管 (initial lymphatics) 亦已存在嚴重的結構與功能變化。³² 法布瑞氏病病人已被鑑定出血清中血管內皮生長因子 (vascular endothelial growth factor, VEGF)-A 濃度顯著升高;VEGF-A 是一種已知會刺激內皮細胞遷移與增生,並增加微血管通透性與血管舒張的醣蛋白。³³
近期鑑定出的淋巴管新生 (lymphangiogenesis) 介質(VEGF-C/-D 與 VEGF-Rs)³⁴ 其表現或功能的異常是否可能參與法布瑞氏病淋巴水腫的發展,仍有待研究。
排汗異常 (ABNORMALITIES OF SWEATING)
排汗減少是法布瑞氏病的典型特徵,一般認為主要是自主神經病變 (autonomic neuropathy) 的後果,儘管汗腺內的受質堆積 (substrate accumulation) 亦可能扮演角色。³⁵,³⁶ 少汗症 (hypohidrosis) 由 53% 的男性與 28% 的女性報告,男性發病較早(平均年齡:23 歲 vs 26 歲)。無汗症 (anhidrosis) 由 25% 的男性描述,但僅 4% 的女性。⁸ 熱不耐受 (heat intolerance) 是常見且令人失能的相關症狀,導致運動耐受度降低、噁心、呼吸困難、頭暈、頭痛,或完全虛脫並喪失意識。先前的研究³⁶,³⁷ 證明接受酵素替代療法 (enzyme replacement therapy, ERT) 的法布瑞氏病病人排汗有所改善。多汗症 (hyperhidrosis) 也可能發生,且女性較男性更常見:369 名女性中有 44 名(11.9%),相對於 345 名男性中有 22 名(6.4%)。³⁸ 此比例高於美國一般族群估計的 1.0% 至 2.8% 盛行率。³⁹ 在多數法布瑞氏病病人中,多汗症影響手掌與足底,並非全身性。³⁸
雷諾現象 (RAYNAUD PHENOMENON)
「冷不耐受」(“Cold intolerance”) 以及在寒冷環境中四肢疼痛的發展,是法布瑞氏病病人常見的主訴。登錄資料記載,雷諾現象見於 710 名女性中的 57 名(8%)與 644 名男性中的 71 名(11%)(資料來自法布瑞結局調查 Fabry Outcome Survey)。¹⁴ 兩項較新的研究鑑定出雷諾現象見於 15.3% 至 38% 的法布瑞氏病病人。⁴⁰,⁴¹ 此比例顯著高於一般族群報告的 3% 至 22% 盛行率,在一般族群中男性受影響的比例(0.5% 至 16%)低於女性(2.5% 至 22%)。⁴¹ 性別比例的逆轉與男性的高盛行率,提示其可能與潛在的法布瑞氏病有因果關聯。在一項以螢光透視甲褶微血管鏡 (fluoroscopic nailfold capillaroscopy) 檢查 25 名法布瑞氏病病人(17 名男性)的研究中,⁴² 病例組(72%)較對照組(10%)鑑定出顯著更多的灌叢狀微血管 (bushy capillaries) 與簇集 (clusters)。甲褶微血管存在型態與功能上的異常。在另一項針對 32 名法布瑞氏病病人(其中 25 名 [78%] 正接受 ERT)的微血管鏡研究中,唯一鑑定出的顯著差異為微血管分支 (ramification) 的增加。⁴¹ 雖然指端血管的異常血管反應性 (vasoreactivity) 可能與自主神經功能障礙有關,但一氧化氮合成酶 (nitric oxide synthetase) 的異常,以及血管內皮與平滑肌氧化壓力 (oxidative stress) 的增加,也可能是重要的誘發因素。⁴³
非皮膚表現 (NONCUTANEOUS FINDINGS)
法布瑞氏病的非皮膚表現如圖 127-2 所示,並依其典型發病年齡列於表 127-1。法布瑞氏病是一種進行性、多系統的疾病,典型導致受影響個體生活品質的全面下降。兒童期的症狀典型與嗜睡、疲倦、疼痛、皮膚異常、感覺器官變化,以及常見的胃腸道障礙有關。在成年早期,病人可能出現上述任何症狀的擴展,並常發展出淋巴水腫、蛋白尿,以及腎臟、心臟或中樞神經系統/腦血管疾病的最初徵象。在成年晚期(年齡 >30 歲),症狀包括上述情況的惡化,以及更嚴重的器官功能障礙(心臟疾病、腎臟疾病與腦血管疾病)。
神經學發現 (NEUROLOGIC FINDINGS)
肢端感覺異常 (Acroparesthesias) 發生於 80% 至 90% 的受影響個體,且典型出現於第一個十年。¹⁶
表 127-1 內容(非皮膚表現):
- 腎臟 (Renal):慢性腎衰竭 (Chronic renal failure)
- 心血管 (Cardiovascular):高血壓 (Hypertension)、冠狀血管疾病 (Coronary vascular disease)、左心室肥大 (Left ventricle hypertrophy)、瓣膜疾病 (Valve disease)、心律不整 (Arrhythmias)、高密度脂蛋白升高 (Elevated high-density lipoprotein)
- 神經學/精神科 (Neurologic/psychiatric):肢端感覺異常 (Acroparesthesias)、腦血管疾病 (Cerebrovascular disease)、失智 (Dementia)、憂鬱 (Depression)、自殺意念增加 (Increased suicide ideation)
- 眼科 (Ocular):渦狀角膜 (Cornea verticillata)、結膜血管動脈瘤 (Conjunctival vessel aneurysms)、視網膜血管迂曲增加 (Increased tortuosity of retinal vessels)、法布瑞白內障 (Fabry cataract)
- 胃腸道 (GI):腹瀉 (Diarrhea)、噁心 (Nausea)、嘔吐 (Vomiting)、餐後脅腹痛 (Postprandial flank pain)、吸收不良 (Malabsorption)
- 其他 (Others):聽力喪失與耳鳴 (Hearing loss and tinnitus)、甲狀腺功能低下 (Hypothyroidism)、阻塞性氣道疾病 (Obstructive airway disease)、骨質缺乏與骨質疏鬆 (Osteopenia and osteoporosis)、貧血 (Anemia)
病人將此感覺描述為疼痛,感覺像手腳針刺感 (pins and needles),常向近端放射。誘發因素包括體溫升高、運動與壓力。疼痛通常隨時間下降,這可能是發炎與受質堆積導致神經纖維受損的結果。
感覺器官異常 (SENSORY ORGAN ABNORMALITIES)
最常見的眼部發現是渦狀角膜 (cornea verticillata,角膜混濁,特徵為一條或多條由角膜中心附近向外放射的線條),見於超過 90% 的法布瑞氏病男性與 70% 的法布瑞氏病男性。其他變化包括視網膜血管迂曲增加、視神經萎縮 (optic atrophy)、白內障與水晶體變化 (lenticular changes)。眼部異常的程度與疾病的整體程度相關。⁴⁴ 眼部變化亦與基因型相關,在帶有無義突變 (nonsense mutations) 的個體中較為嚴重。⁴⁵ 耳鳴與高頻感覺神經性聽力喪失 (high-frequency sensorineural hearing loss) 也是常見表現,發生於超過 50% 的病人。¹⁶
由前庭病理 (vestibular pathology) 所致的突發性耳聾與眩暈亦可能發生。¹⁶
胃腸道變化 (GASTROINTESTINAL CHANGES)
絞痛性腹痛、噁心、腹瀉以及偶爾便祕都很常見,且常是表現症狀。⁴⁶ 其發病機轉可能與神經學異常有關。
法布瑞氏病的器官損傷 (ORGAN DAMAGE IN FABRY DISEASE)
腎臟表現 (RENAL MANIFESTATIONS)
腎臟表現見於超過 90% 的男性。¹⁶ 微白蛋白尿 (Microalbuminuria) 與超過濾 (hyperfiltration) 是早期特徵;蛋白尿典型見於第三與第四個十年;並會發生腎臟過濾能力的進行性下降。女性常出現蛋白尿,但進展至末期腎衰竭 (end-stage renal failure) 較少見。一種法布瑞氏病的腎臟變異型已被描述於 α-galactosidase A 活性降低但非完全缺失的病人;¹ 這些病人缺乏其他特徵性表現。
心臟表現 (CARDIAC MANIFESTATIONS)
心臟表現是一恆定特徵,且日益被認為是男女病人的主要死因。⁴⁷ 受質沉積可在整個心肌、瓣膜與傳導系統中顯示,且常伴隨發炎細胞浸潤。常見的表現為左心室肥大 (left ventricular hypertrophy),但二尖瓣脫垂 (mitral valve prolapse)、心律不整與冠狀動脈疾病皆可能出現。一種心臟變異型¹ 已被描述於 α-galactosidase A 活性降低但非完全缺失的病人,且於較晚年發病(常於病人年齡大於 40 歲時),以心臟症狀為主。磁振造影 (MRI) 掃描在偵測早期侵犯,以及顯示纖維化 (scarring,為晚期疾病的特徵) 方面具有寶貴的作用。⁴⁸
腦血管表現 (CEREBROVASCULAR MANIFESTATIONS)
腦血管表現包括早年發生的缺血性或出血性中風,以及短暫性腦缺血發作與影響後循環 (posterior circulation) 的中風。中風常被報告為法布瑞氏病的表現特徵,且對於罹患隱源性中風 (cryptogenic stroke) 的年輕病人,鑑別診斷應將法布瑞氏病納入考量。⁴⁹ 曾中風的法布瑞氏病病人中,第五凝血因子萊登突變 (factor V Leiden) 的盛行率增加。⁵⁰ 在年紀較大的法布瑞氏病男女病人中可顯示認知功能障礙。⁵¹
其他表現 (OTHER MANIFESTATIONS)
法布瑞氏病的整體表現包括嗜睡、疲倦、兒童發育不良 (failure to thrive) 與貧血。憂鬱很常見且常被低估診斷,影響多達 50% 的法布瑞氏病男女病人。⁵² 生殖器區域血管角化瘤的存在常影響性活動,並可能導致自尊與性慾降低。自主神經功能障礙未被充分認識,可能導致多汗症、淚液與唾液分泌異常、異常的心臟反應性、胃腸道蠕動障礙 (dysmobility)、痛覺與溫度感覺改變,以及周邊水腫。內分泌異常不常見,但骨質缺乏與甲狀腺功能低下⁵³ 已被充分描述。肺部異常未被充分認識;法布瑞氏病在某些病人可導致阻塞性氣道模式 (obstructive airways pattern),⁵⁴ 但在其他病人氣喘是其表現特徵。與其他溶體儲積症不同,認知功能障礙一般不會出現;然而,年紀較大的病人(年齡大於 50 歲)愈來愈多出現記憶喪失、整體智力退化與人格改變。⁵¹
診斷 (DIAGNOSIS)
法布瑞氏病的確定診斷通常會延遲,從症狀發作到診斷之間的平均時間為 15 年。⁵⁵ 診斷必須藉由證明血漿、血清或白血球中 α-galactosidase A 活性缺乏,並鑑定致病性突變來確認。女性病人的酵素活性程度變異很大,偶爾甚至正常,使得 DNA 確認診斷成為必要。皮膚與腎臟等組織的切片,於電子顯微照片中顯示脂質沉積與多層髓鞘小體 (multilamellated myelin bodies)(見圖 127-7)。由於 Gb3 沉積始於子宮內 (in utero),可從絨毛膜絨毛 (chorionic villi) 或羊水細胞 (amniotic cells) 培養進行產前診斷 (prenatal diagnosis),可顯示低 α-galactosidase A 活性。¹ 晚發型突變的重要性日益受到認識;確認突變的致病性 (pathogenicity) 非常重要。測量三己醣鞘氨醇 (globotriaosylsphingosine) 可增加有價值的資訊。⁵⁶
鑑別診斷 (DIFFERENTIAL DIAGNOSIS)
與肢端感覺異常相關的疼痛常被誤診為類風濕性關節炎 (rheumatoid arthritis)、風濕熱 (rheumatic fever)、紅斑性肢痛症 (erythromelalgia)、雷諾氏病 (Raynaud disease),或單純被當作「生長痛」(“growing pains”)。¹⁶
表 127-2 內容(血管角化瘤的鑑別診斷):
- 局限型 (LOCALIZED FORMS):佛代斯血管角化瘤 (Angiokeratoma of Fordyce)、外陰血管角化瘤 (Angiokeratoma of the vulva)、米貝利血管角化瘤 (Angiokeratoma of Mibelli)、單發丘疹型血管角化瘤 (Solitary papular angiokeratoma)、限界型血管角化瘤 (Angiokeratoma circumscriptum)
- 瀰漫性軀體血管角化瘤 (ANGIOKERATOMA CORPORIS DIFFUSUM):岩藻糖苷貯積症 (Fucosidosis)、天門冬醯胺葡萄糖胺尿症 (Aspartylglycosaminuria)、半乳唾液酸貯積症 (Galactosialidosis)、Schindler/Kanzaki 病、β-甘露糖苷貯積症 (β-Mannosidosis)、GM1 神經節苷脂貯積症/β-半乳糖苷酶缺乏 (GM1-gangliosidosis/β-galactosidase)、唾液酸貯積症 (Sialidosis)、特發性 (Idiopathic)
法布瑞氏病中血管角化瘤的臨床診斷可能有困難。可能需要仔細檢查皮膚,以將其與紫斑 (purpura)、瘀點 (petechiae) 與匐行性血管瘤 (angioma serpiginosum) 區分。也應將其與在無潛在系統性疾病情況下發生的單發及局限型血管角化瘤區分(表 127-2 與表 127-3)。⁵⁷
廣泛性血管角化瘤亦發生於其他溶體儲積症,應納入鑑別診斷考量。這些包括岩藻糖苷貯積症 (fucosidosis),⁵⁸ 其中超過 50% 的病人有此變化、α-N-半乳糖胺酶缺乏症 (α-N-galactosaminidase deficiency) 與半乳唾液酸貯積症 (galactosialidosis)(47% 至 50%)(見表 127-2 與表 127-4)。除了這些貯積症各自獨特的酵素缺乏與其他臨床特徵外,法布瑞氏病的血管角化瘤可藉由電子顯微鏡檢查,依據內皮細胞與其他細胞類型內特徵性的電子緻密、層狀(斑馬狀)內含物加以區別。⁵⁹⁻⁶² 氯奎寧 (Chloroquine) 治療可能導致生化與超微結構上類似的內含物儲積於許多與法布瑞氏病相同的細胞中,並可能導致類似的臨床表現,包括血管角化瘤的發展。⁶³ 廣泛性血管角化瘤亦曾被描述與結節性硬化症 (tuberous sclerosis)、⁶⁴ 幼年型皮肌炎 (juvenile dermatomyositis)⁶⁵ 相關聯,以及在無相關代謝疾病的情況下發生。⁶⁶
預後與臨床病程 (PROGNOSIS AND CLINICAL COURSE)
較早的研究指出,法布瑞氏病男性典型在第四或第五個十年死亡,女性則或許多活約 15 年。較近期,男性的預期壽命估計為 58.2 歲(相較於美國一般族群的 74.7 歲),女性為 75.4 歲(相較於一般族群的 80 歲)。⁶⁷ 亦有證據顯示,雖然腎衰竭先前是最常見的死因,但心臟疾病與腦血管疾病正日益常見。這些變化部分可能是 ERT 影響的結果。⁶⁷,⁶⁸
表 127-3 內容(不同類型血管角化瘤的臨床特徵;欄位:類型 TYPE/發病年齡(歲)AGE AT ONSET (YR)/性別 SEX/臨床面向 CLINICAL ASPECT/身體部位 BODY SITE/遺傳 INHERITED/關聯 ASSOCIATION):
類型 發病年齡(歲) 性別 臨床面向 身體部位 遺傳 關聯 法布瑞 (Fabry) 5-12 兩性,男性 > 女性 多發、群聚的暗紅色至藍黑色斑疹與丘疹;較大病灶呈疣狀 (warty)(1-4 mm) 身體任何部位,尤其泳褲區、肚臍、嘴唇 是,X 染色體連鎖 肢端感覺異常、心臟與腎衰竭、中風、渦狀角膜、耳聾等 米貝利 (Mibelli) 10-15 兩性,女性 > 男性 群聚、疣狀、暗紅色丘疹(1-5 mm) 手指與足趾的外側面與背面、手與足 是,自體顯性 肢端發紺 (acrocyanosis) 與凍瘡 (chilblains) 佛代斯 (Fordyce) >60 男性 多發疣狀、藍黑色丘疹(1-4 mm) 生殖器,主要為陰囊 否 局部靜脈高壓 外陰 (Vulva) >60 女性 群聚、疣狀、藍黑色丘疹(1-4 mm) 外陰 否 局部靜脈高壓 單發丘疹型 (Solitary papular) 10-40 兩性 單一暗紅色至黑色角化性丘疹(2-10 mm),常出血/血栓化 身體任何部位,尤其下肢 否 — 限界型 (Circumscriptum) 通常早發或出生時 兩性 單側斑塊狀角化性暗紅色丘疹,可能出血;下肢或足部可能呈帶狀分布 (zosteriform)⁵³ 下肢或足部 否 Cobb 症候群;血管畸形 (vascular malformations)
治療 (TREATMENT)
表 127-4 內容(瀰漫性軀體血管角化瘤的鑑別診斷;欄位:族群 GROUP/名稱 NAME/酵素缺乏 ENZYME DEFICIENCY/基因 GENE/電子顯微鏡發現 ELECTRON MICROSCOPY FINDINGS⁵⁵/臨床發現 CLINICAL FINDINGS/皮膚科發現 DERMATOLOGIC FINDINGS):
族群 名稱 酵素缺乏 基因 電子顯微鏡發現⁵⁵ 臨床發現 皮膚科發現 鞘脂質貯積症 (Sphingolipidoses) 法布瑞 Fabry (OMIM #301500) α-Galactosidase A Xq22 電子緻密溶體沉積 (Electron-dense lysosomal deposits) 肢端感覺異常、心臟與腎衰竭、中風、渦狀角膜 瀰漫性軀體血管角化瘤、毛細血管擴張、少汗症/無汗症、淋巴水腫 — GM1 神經節苷脂貯積症 GM1-gangliosidosis (OMIM #230500) β-Galactosidase 3p21-3pter 電子透亮溶體擴張 (Electron-lucent lysosomal dilation) 面部畸形、血液學徵象、智能障礙、器官腫大 瀰漫性軀體血管角化瘤 醣蛋白貯積症 (Glycoproteinoses) 天門冬醯胺葡萄糖胺尿症 Aspartylglucosaminuria (OMIM #208400) Aspartylglycosaminidase 4q32-33 電子透亮溶體擴張 粗獷面容、巨舌 (macroglossia)、器官腫大、眼部發現、心臟瓣膜侵犯 瀰漫性軀體血管角化瘤、面部血管纖維瘤 (facial angiofibromas)、口腔纖維瘤病 (oral fibromatosis) 與白色角化症 (leukokeratosis) — 岩藻糖苷貯積症 Fucosidosis (OMIM #230000) α-Fucosidase 1p34 電子透亮溶體擴張 智能障礙、粗獷面容、生長遲緩、反覆呼吸道感染、多發性骨發育不全 (dysostosis multiplex)、內臟腫大 瀰漫性軀體血管角化瘤、廣泛毛細血管擴張、肢端發紺、紫色橫向遠端甲帶、手足血管增生、排汗異常 — β-甘露糖苷貯積症 β-Mannosidosis (OMIM #248510) β-Mannosidase 4q22-q25 電子透亮溶體擴張 智能障礙、神經病變、聽力喪失、反覆感染 泳褲區的瀰漫性軀體血管角化瘤 — 唾液酸貯積症 II 型 Sialidosis II (OMIM #256550) Neuraminidase 6p21.3 電子透亮溶體擴張 智能障礙、多發性骨發育不全、空泡化淋巴球、輕微粗獷面容 瀰漫性軀體血管角化瘤 多重酵素缺乏 (Multiple enzyme deficiency) 半乳唾液酸貯積症 Galactosialidosis (OMIM #256540) β-Galactosidase 與 neuraminidase 20q13.1 電子透亮溶體擴張 侏儒症 (Dwarfism)、滴水嘴獸面容 (gargoyle facies)、智能障礙、癲癇、角膜混濁、多發性骨發育不全與聽力喪失 散布於全身的瀰漫性軀體血管角化瘤,尤其膝、肘與泳褲區 — Kanzaki (OMIM #104170) α-N-acetylgalactosaminidase 22q11 電子透亮溶體擴張 智能障礙、粗獷面容、眼部徵象、聽力喪失、神經病變 全身性瀰漫性軀體血管角化瘤,於泳褲區、腋下與乳房較密集;嘴唇與口腔黏膜毛細血管擴張 OMIM,線上人類孟德爾遺傳 (Online Mendelian Inheritance in Man)。
ERT 的問世意味著病人與家屬日益於專科中心接受評估,在此他們可接觸到內科醫師或小兒科醫師、皮膚科醫師,以及可能的心臟科/腎臟科/神經科醫師。在較大型的中心,也能接觸並提供聽力師、眼科醫師、腸胃科醫師、精神科醫師/諮商師,以及遺傳諮詢師與護理人員的服務。
症狀治療 (SYMPTOMATIC THERAPIES)
個別末端器官的疾病表現以症狀方式治療(見表 127-5)。
皮膚導向治療 (SKIN-DIRECTED THERAPIES)
雖然有報告指出 ERT 後皮膚的 Gb3 清除,⁶⁹,⁷⁰ 但這不必然轉化為血管角化瘤的清除。然而,法布瑞氏病所見的各種血管病灶可用準連續波 (quasicontinuous wave) 與脈衝雷射 (pulsed lasers),以及脈衝光系統 (intense pulsed light systems) 有效治療。⁷¹,⁷² 較新的雷射結合了高功率脈衝染料雷射 (high-powered pulsed-dye laser) 與 1064-nm 長脈衝 Nd:YAG 雷射,能穿透更深,似乎對較大的生殖器血管角化瘤特別有效。⁷³,⁷⁴
對於水腫仍完全可逆的病人,以第二級漸進式膝下壓力襪 (grade II graduated below-knee compression hosiery) 進行預防性治療,可預防淋巴水腫的發展。一旦淋巴水腫已形成,可藉由規律的皮膚護理、運動、徒手淋巴引流與/或自我按摩,以及使用適當的專科繃帶與淋巴水腫襪,在多達 80% 的病人中維持控制。⁷⁵
有些證據顯示少汗症可藉由 ERT 改善。對於有多汗症的病人,治療選項包括使用局部六水合氯化鋁 (aluminium chloride hexahydrate)、自來水離子導入 (tap water iontophoresis) 與局部肉毒桿菌毒素 (botulinum toxin) 注射、口服格隆溴銨鈉 (glycopyrrolate sodium) 至每日三次每次 2 mg(一種耐受性良好、副作用極少的抗膽鹼藥物,前提是無心臟禁忌症),以及化學性或內視鏡性交感神經切除術 (sympathectomy)。⁷⁶,⁷⁷
對於四肢冰冷的病人,一般建議採取標準措施,如戒菸、避免使用血管收縮藥物(如乙型阻斷劑 beta-blockers),以及在冬季月份以合適衣物保持手腳溫暖。在其他病人族群中對雷諾現象已證實有效的藥物療法,包括血管收縮素 II 受體拮抗劑 (angiotensin II receptor antagonists)、鈣離子通道阻斷劑 (calcium channel blockers)、氟西汀 (fluoxetine) 與西地那非 (sildenafil)。⁷⁸,⁷⁹ 在為法布瑞氏病病人開立這些藥物前,應考量可能的心臟與腎臟禁忌症。
針對其他器官系統的治療 (THERAPIES DIRECTED AT OTHER ORGAN SYSTEMS)
疼痛是最令人困擾且早期的症狀,有時可用苯妥英 (diphenylhydantoin)、卡馬西平 (carbamazepine) 或加巴噴丁 (gabapentin) 部分控制。應鼓勵病人辨識並嘗試避免其個人的誘發因素。為了中風與其他血管病理(如視網膜動脈阻塞 retinal artery occlusion)的初級或次級預防,可能需要抗血小板 (antiplatelet) 與抗凝血 (anticoagulant) 治療。甲氧氯普胺 (Metoclopramide) 與胰脂酶 (pancrelipase) 用於減輕胃腸道症狀。高血壓必須控制,因為它顯著影響三個最常受影響的器官:腎臟、腦與心臟。應在出現蛋白尿的最初徵象時即進行血管收縮素受體阻斷 (angiotensin-receptor blockade),這是 ERT 在減緩腎功能下降方面的重要輔助。法布瑞氏病病人在發生末期腎衰竭時是良好的腎臟移植候選者,而 ERT 在此情況下的作用是保存其他器官的功能。心臟方面的輔助照護包括考慮抗心律不整藥物、人工心律調節器,以及手術,包括中膈消融術 (septal ablation) 與心臟移植。
特異性治療:酵素替代療法 (SPECIFIC THERAPEUTICS: ENZYME REPLACEMENT THERAPY)
已開發出兩種 ERT 製劑:agalsidase beta(Genzyme)與 agalsidase alpha(Replagal)。在美國僅 agalsidase beta 獲核准,而在世界其他大部分地區兩種製劑皆可取得。Agalsidase beta 以 1 mg/kg 每兩週一次 (biweekly) 的劑量給予,並使用重組技術 (recombinant technology) 於中國倉鼠卵巢細胞株 (Chinese hamster ovary cell line) 製造。Agalsidase alpha 以 0.2 mg/kg 每兩週一次的劑量給予,並使用基因活化方法 (gene activation methodology) 於人類纖維母細胞株 (human fibroblast cell line) 製造。兩種 ERT 製劑的療效已於隨機對照試驗中得到證明,⁸⁰⁻⁸² 且兩者皆改善生化(例如血漿、尿液與組織切片中的 Gb3 濃度)與臨床參數。所選用於研究的主要臨床參數為腎功能、疼痛、心臟大小與功能,以及生活品質。⁸³ 兩種製劑的長期有效性(具超過 10 年的資料)已於登錄研究中得到證明。⁸⁴,⁸⁵ 兩種製劑皆被認為安全且耐受性良好。主要副作用與輸注相關(發燒、體溫升高),且兩種製劑皆可能誘發抗體形成。然而,抗體對臨床有效性的影響尚未被證明。當以其核准劑量使用時,這兩種 ERT 製劑一般被認為具有同等的有效性。⁸⁶ 開始治療的最佳時機尚未確立。建議所有有症狀的男性,以及女性在出現器官功能障礙的最初徵象時,接受 ERT。接受 ERT 的病人應以連續測量疼痛、生活品質,以及腎臟與心臟功能進行規律監測。其資料應盡可能輸入登錄資料庫。⁸⁷ ERT 可改善聽力與胃腸道症狀。ERT 對中樞神經系統異常的直接有益效果尚未被證明,且酵素無法通過血腦障壁 (blood–brain barrier)。
新療法 (NEW TREATMENTS)
以伴護蛋白為基礎的酵素增強療法 (Chaperone-based enzyme enhancement therapy) 由小分子組成,這些小分子可從溶體中拯救折疊錯誤/運輸錯誤 (misfolded/mistrafficked) 的酵素,並將其運送至內質網 (endoplasmic reticulum)。以伴護蛋白為基礎的療法可口服給予,並已在帶有可受惠突變 (amenable mutations) 與殘餘活性的個體中顯示活性。⁸⁸ 米格司他 (Migalastat) 已獲美國與歐洲監管機構核准。受質剝奪 (Substrate deprivation,抑制醣鞘脂質合成中的早期步驟) 與半乳糖 (galactose) 輸注是仍處於研究階段的其他治療選項。基因療法 (Gene therapy) 與造血細胞移植 (hematopoietic cell transplantation) 仍在開發中。
遺傳諮詢 (GENETIC COUNSELING)
所有有成員罹患法布瑞氏病的家族都應有機會接受遺傳諮詢。鑑於法布瑞氏病的 X 染色體連鎖性質,女性病人有 50% 的機率將基因傳給其兒子與女兒,而男性病人不會有受影響的兒子,但 100% 的女兒會受影響。亦應強調基因型—表型 (genotype–phenotype) 的變異性,尤其在有輕微表現的病人中。
圖表 (Figures and Tables)
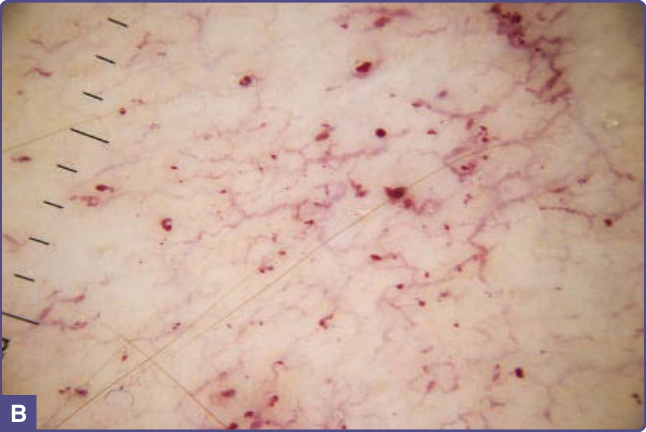
圖 127-1:三己醣神經醯胺 (globotriaosylceramide) 的代謝路徑。α-半乳糖苷酶 A (α Gal A) 將三己醣神經醯胺代謝為半乳糖 (Gal, galactose) 與乳醣神經醯胺 (lactosylceramide)。Cer,神經醯胺 (ceramide);Glu,葡萄糖 (glucose)。
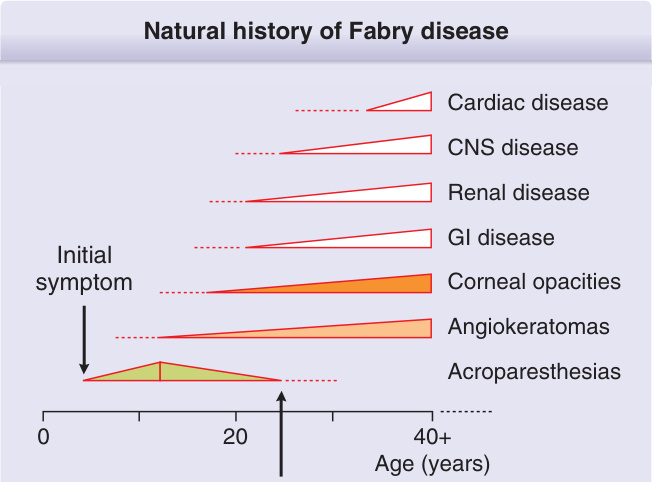
圖 127-2:法布瑞氏病的自然病程。肢端感覺異常 (acroparesthesias)、血管角化瘤 (angiokeratomas)、角膜混濁 (corneal opacities) 與胃腸道症狀常為最初表現。診斷通常延遲,約於第二個十年發生。
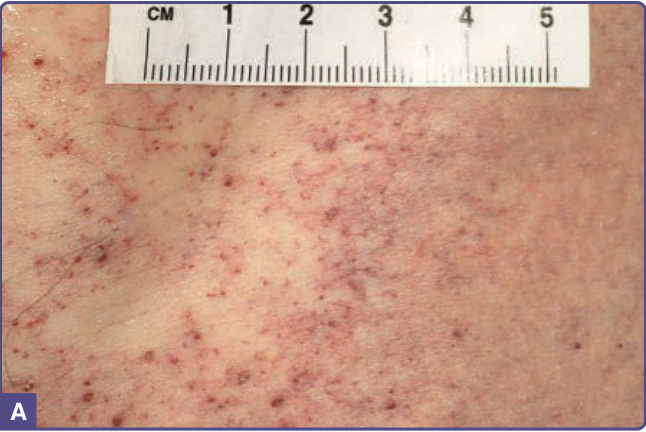
圖 127-3:A,一名瀰漫性軀體血管角化瘤 (angiokeratoma corporis diffusum) 病人脅腹 (flank) 上的血管角化瘤與毛細血管擴張血管 (telangiectatic vessels)。B,皮膚鏡 (dermatoscopic) 影像顯示血管角化瘤與真皮上層血管迂曲 (upper dermal vessel tortuosity)。
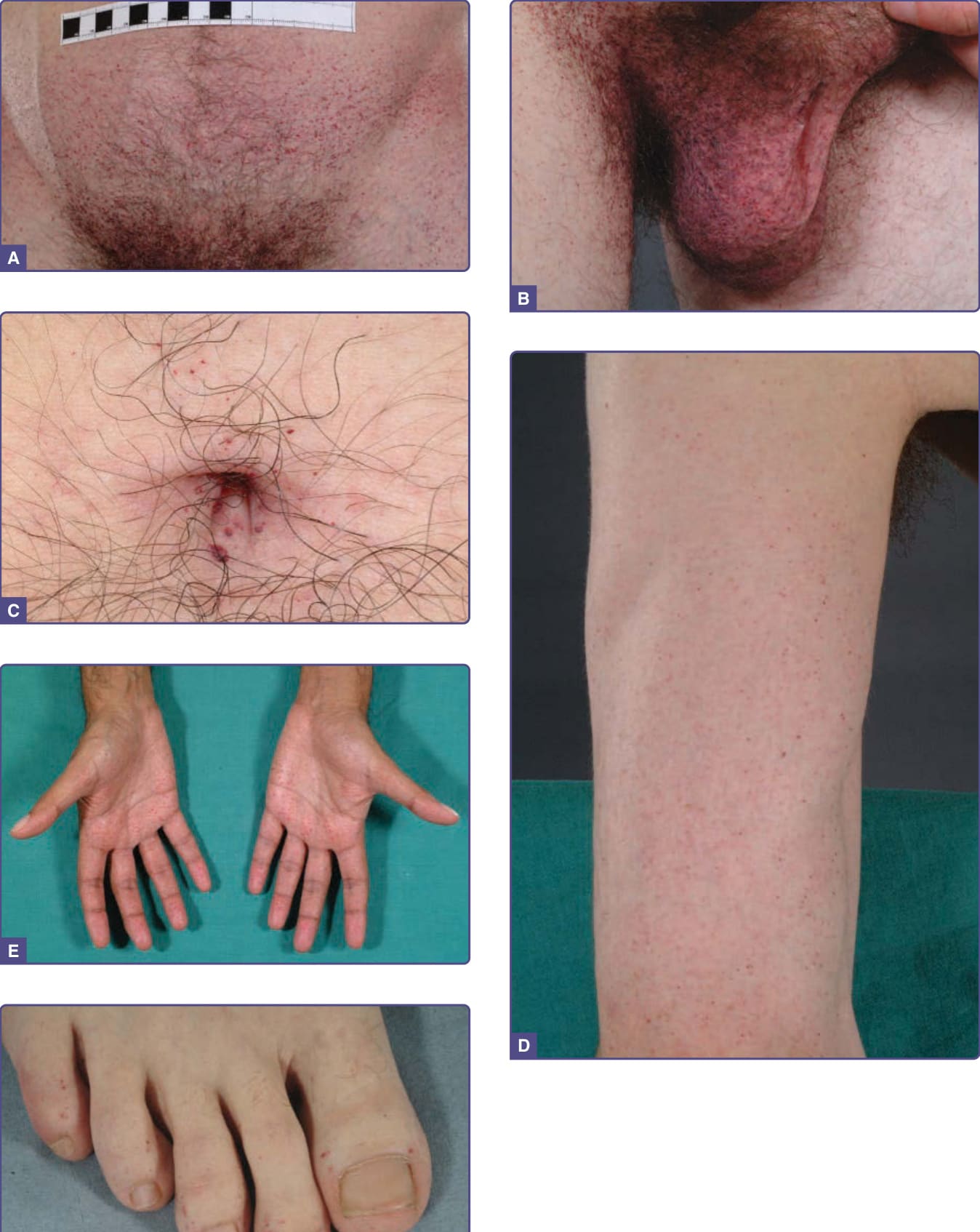
圖 127-4:疾病完全表型表現之法布瑞氏病男性的血管角化瘤分布。A,下腹部;B,陰囊與大腿內側;C,肚臍;D,上肢;E,手掌;F,足趾。
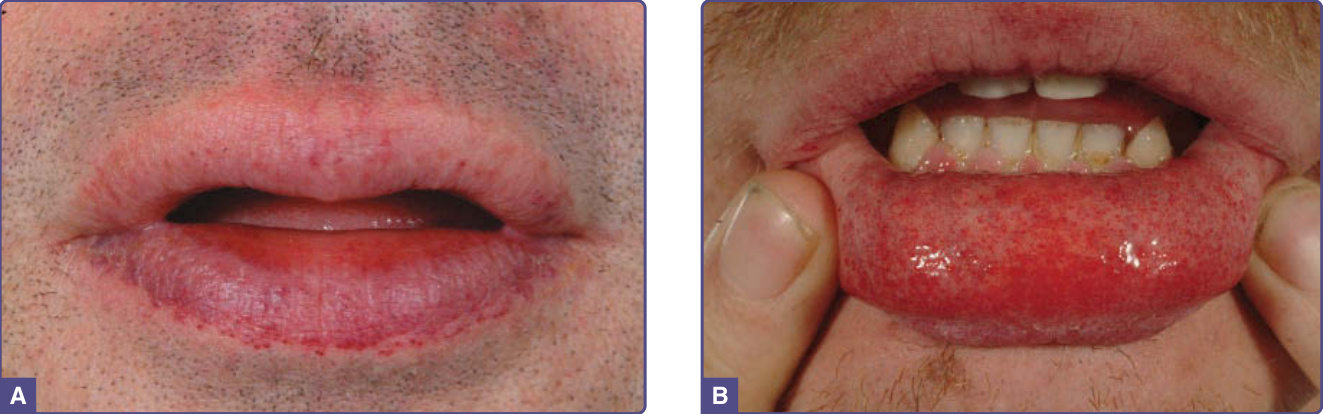
圖 127-5:嘴唇上的血管瘤性病灶 (angiomatous lesions)。A,集中於唇紅緣 (vermilion border);B,於下唇黏膜表面。
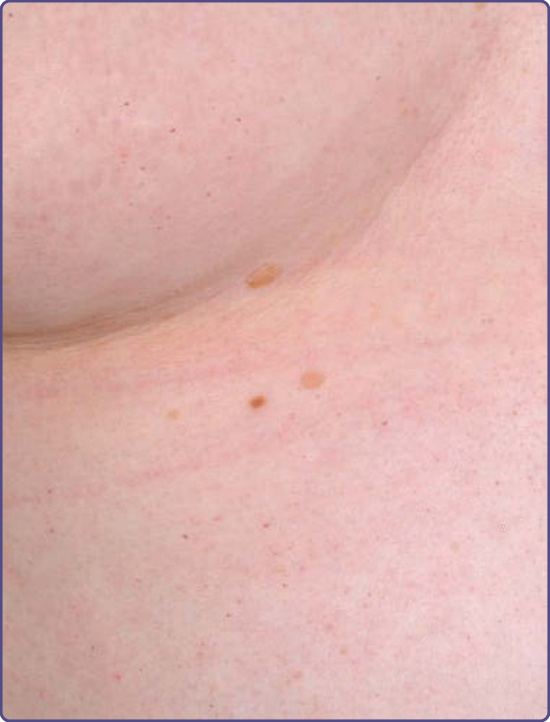
圖 127-6:一名法布瑞氏病女性軀幹上稀疏分布的針尖大小暗紅色斑疹與丘疹病灶。
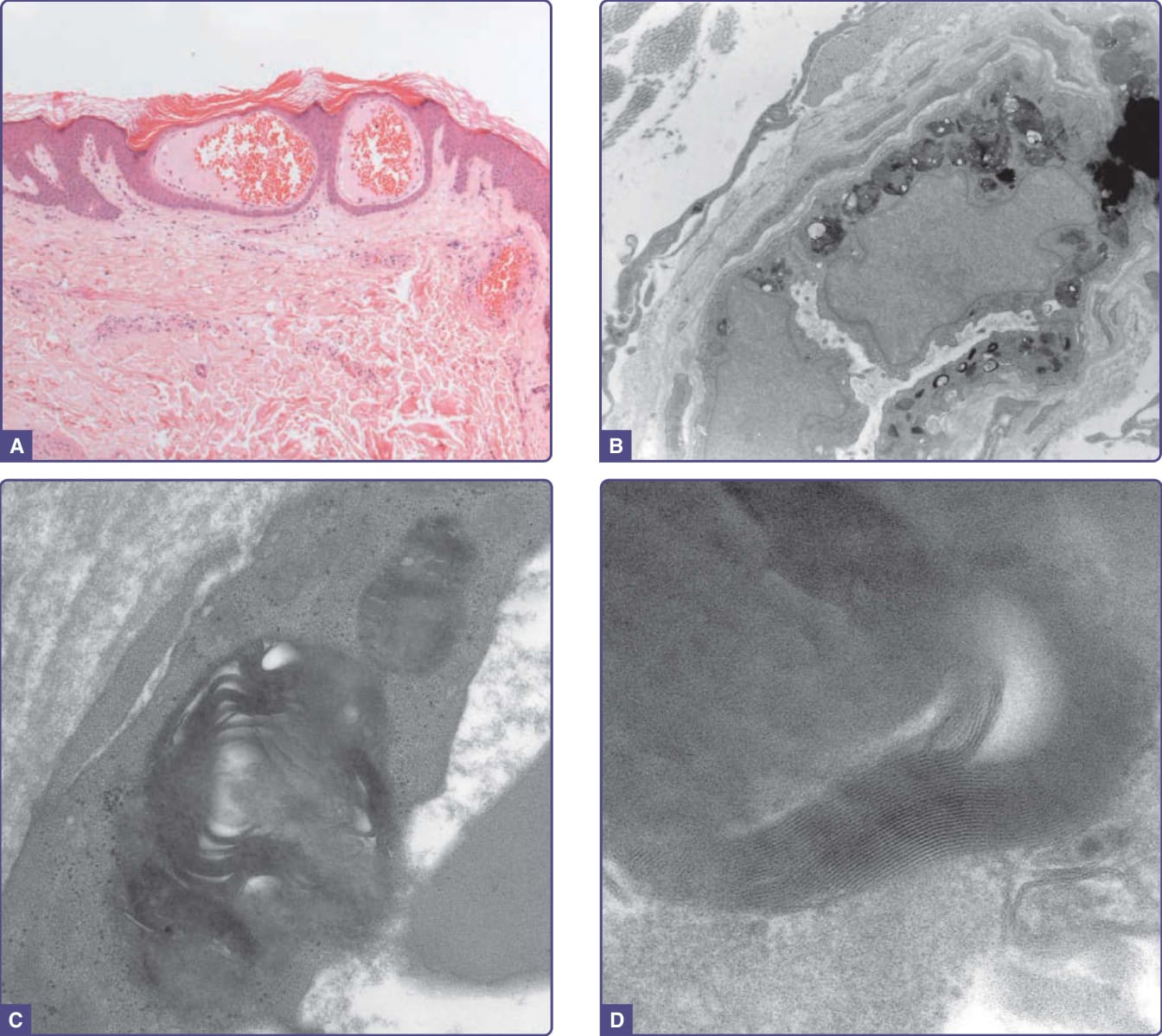
圖 127-7:血管角化瘤的組織病理學特徵。A,光學顯微鏡 (light microscopy)。真皮乳頭層 (papillary dermis) 內擴張的微血管,其上方表皮變薄並有輕度正角化過度 (orthohyperkeratosis)。周邊表皮棘層增厚 (acanthotic epidermis)。蘇木精與伊紅 (hematoxylin and eosin, H&E) 染色,×40 倍放大。B-D,電子顯微鏡 (electron microscopy)。B,皮膚血管內皮細胞內多個電子緻密內含物(×9000 倍放大)。C,於較高倍率下可見一層狀或「斑馬」(“zebra”) 小體(×63,000 倍放大)。D,於最高倍率下(×223,000 倍放大)可辨識出週期性 4 至 6 nm 的明暗條帶。
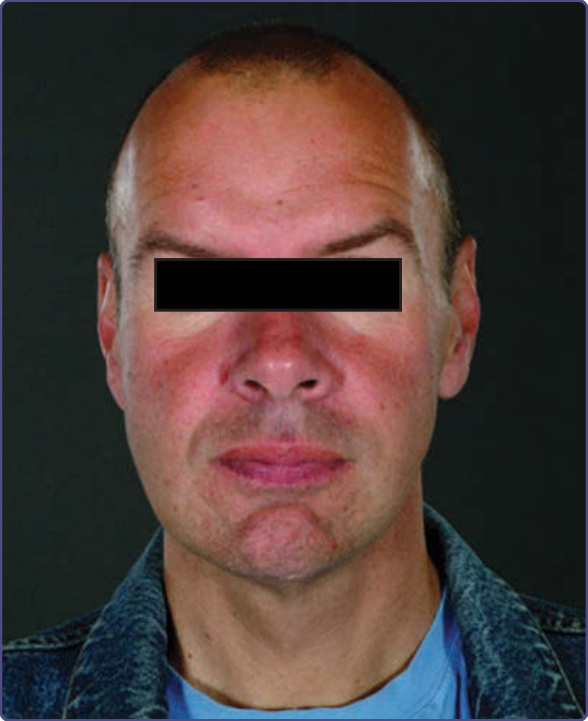
圖 127-8:典型面部特徵:突出的眶上脊 (supraorbital ridges)、眼周飽滿 (periorbital fullness)、寬大的雙顳寬度 (bitemporal width)、濃眉 (bushy eyebrows)、寬闊的鼻基部、豐厚的嘴唇,以及突出的下巴。
圖 127-9:一名 43 歲法布瑞氏病男性的雙側下肢淋巴水腫 (bilateral lower-limb lymphedema)。

表 127-1:非皮膚表現 (Noncutaneous Findings)

表 127-2:血管角化瘤的鑑別診斷 (Differential Diagnosis of Angiokeratomas)
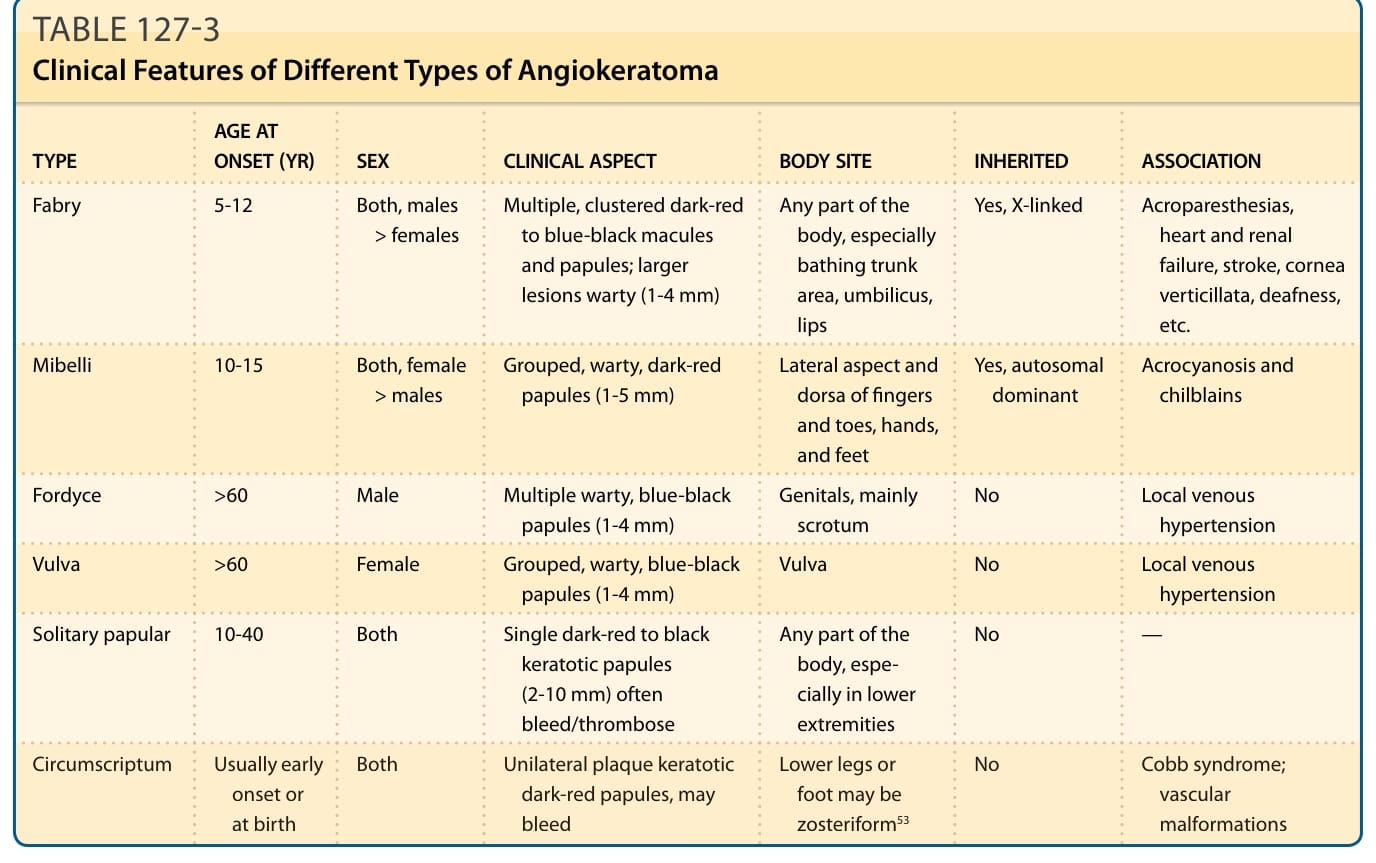
表 127-3:不同類型血管角化瘤的臨床特徵 (Clinical Features of Different Types of Angiokeratoma)
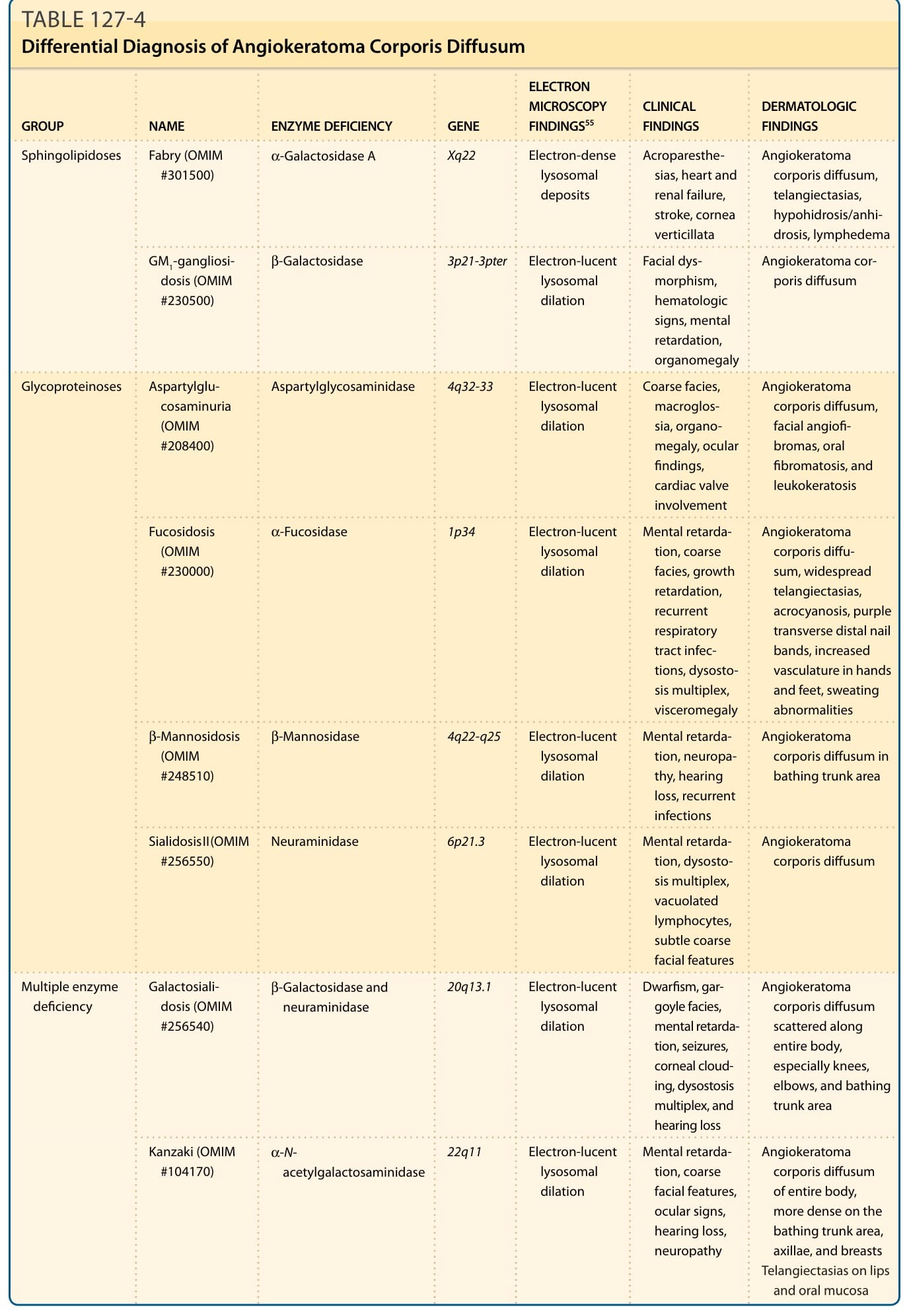
表 127-4:瀰漫性軀體血管角化瘤的鑑別診斷 (Differential Diagnosis of Angiokeratoma Corporis Diffusum)
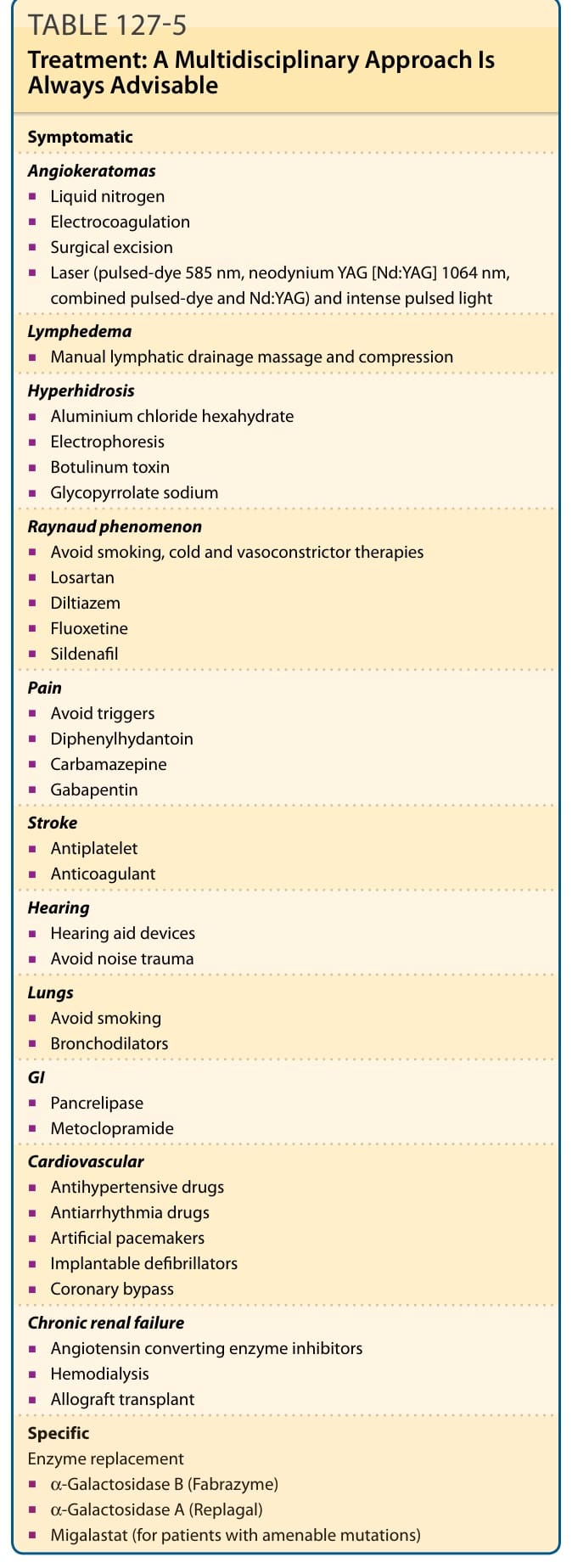
表 127-5:本表概述法布瑞氏病的治療 (Fabry disease treatment)。法布瑞氏病是一種多系統疾病,病人應在多專科團隊 (multidisciplinary) 的環境中接受評估。表內治療涵蓋——血管角化瘤 (Angiokeratomas):液態氮、電燒灼術、手術切除、雷射(脈衝染料 585 nm、釹雅鉻雷射 [Nd:YAG] 1064 nm、合併脈衝染料與 Nd:YAG)與脈衝光;淋巴水腫 (Lymphedema):徒手淋巴引流按摩與壓迫;多汗症 (Hyperhidrosis):六水合氯化鋁、電泳 (electrophoresis)、肉毒桿菌毒素、格隆溴銨鈉 (glycopyrrolate sodium);雷諾現象 (Raynaud phenomenon):避免吸菸、寒冷與血管收縮療法、losartan、diltiazem、fluoxetine、sildenafil;疼痛 (Pain):避免誘發因素、苯妥英 (diphenylhydantoin)、卡馬西平 (carbamazepine)、加巴噴丁 (gabapentin);中風 (Stroke):抗血小板、抗凝血;聽力 (Hearing):助聽器裝置、避免噪音創傷;肺部 (Lungs):避免吸菸、支氣管擴張劑;胃腸道 (GI):胰脂酶 (pancrelipase)、甲氧氯普胺 (metoclopramide);心血管 (Cardiovascular):抗高血壓藥物、抗心律不整藥物、人工心律調節器、植入式去顫器、冠狀動脈繞道術;慢性腎衰竭 (Chronic renal failure):血管收縮素轉化酶抑制劑 (ACE inhibitors)、血液透析、同種異體移植。特異性酵素替代 (Specific Enzyme replacement):α-Galactosidase B (Fabrazyme)、α-Galactosidase A (Replagal)、米格司他 (Migalastat,用於帶有可受惠突變之病人)。