Fabry Disease
21
AT-A-GLANCE
■ Incidence estimated at 1:3200 to 1:170,000 population in all ethnicities.
■ X-linked lysosomal storage disorder.
■ Highly penetrant in males; female heterozygotes have variable expressivity.
■ Partial or complete deficiency of α-galactosidase A with deposition of glycosphingolipids (mostly globotriaosylceramide).
■ Classical variants affect predominantly skin, kidneys, heart, eyes, and brain.
■ Life expectancy shortened by 20 years in males and 15 years in females.
■ Later onset variants are milder and predominantly involve a single organ (eg, renal or cardiac).
■ Dermatologic manifestations include angiokeratoma, telangiectases, “pseudoacromegalic” facies, hypohidrosis and hyperhidrosis, lymphoedema, and Raynaud phenomenon.
■ Light microscopy shows ectatic upper dermal vessels, peripheral epidermal acanthosis, and variable hyperkeratosis.
■ Electron microscopy shows intracytoplasmic, electron-dense, vacuolar “Zebra bodies.”
■ Treatment is symptomatic, enzyme replacement; chaperone therapy (for amenable mutations).
EPIDEMIOLOGY
Fabry disease (α-galactosidase A deficiency) is generally considered the second most prevalent lysosomal storage disorder, after Gaucher disease,1,2 with an estimated incidence ranging between 1:40,000 and 1:170,000 persons. All ethnic groups are affected. Neonatal screening studies reveal a higher incidence of mutations in the GLA gene and some of the affected individuals suffer from milder variants of the disease, which are associated with significant residual enzyme activity. These may occur with much higher frequency (eg, 1:3200) as suggested by a recent survey of neonates in Northern Italy.3 A study of more than 7000 neonates in Japan suggests an incidence of pathogenic mutations of approximately 1:70004 and a similar incidence has been reported from Europe.5
These variants often have predominant involvement
of a single organ (eg, heart or kidney) and have onset in adult life. A study of the Taiwan Chinese population revealed an unexpectedly high prevalence of the cardiac-variant Fabry-causing mutation IVS4+919G>A among newborns (approximately 1:1600 males), as well as patients with idiopathic hypertrophic cardiomyopathy.6
ETIOLOGY AND PATHOGENESIS
Fabry disease (Online Mendelian Inheritance in Man #301500) is a rare X-linked metabolic disorder caused by the partial or complete deficiency of a lysosomal enzyme, α-galactosidase A (Fig. 127-1). As a result of this enzyme deficiency, neutral sphingolipids with terminal α-galactosyl residues (predominantly globotriaosylceramide [Gb3]) accumulate in the lysosomes of different tissues and fluids (epithelial cells of glomeruli and tubules of the kidneys; cardiac myocytes; ganglion cells of the autonomic system; cornea; endothelial, perithelial, and smooth muscle cells of blood vessels; and histiocytic and reticular cells of connective tissue). Elevated levels of globotriaosylsphingosine, which is the deacylated derivative of Gb3, correlate with disease severity in males and females. Globotriaosylsphingosine inhibits the activity of α-galactosidase A and it has been suggested that elevated levels in females could be of pathogenic significance.7 Clinical onset is variable (Fig. 127-2). Although the condition is X linked, heterozygous females are frequently affected and may be as severely affected as hemizygous males.8 Clinical symptoms typically occur a decade or so later in females than in males and organ damage in females is usually less severe than in males. Variable expression in females is attributed to the Lyon hypothesis whereby 1 X chromosome is inactivated on a random basis in the female, while the other provides the genetic information. The mutant X chromosome is more likely to be expressed in a range of tissues in symptomatic females with Fabry disease.9
The gene GLA for α-galactosidase A is located at the Xq22.1 region. More than 600 mutations have been identified in the GLA gene,10-12 including missense, nonsense mutations, and single amino acid deletions and insertions. Most of these mutations are “private,” having been identified only in individual families. Some (eg, the p.N215S mutation) are associated with single-organ or late-onset variants and patients have normal levels of urinary Gb3, despite having clinical evidence of Fabry disease.
Metabolic pathway of globotriaosylceramide
α Gal A
Gal Gal α β β Glu
Cer
Gal
Globotriaosylceramide (Gb3)
β β Glu Cer
Gal
Lactosyceramide
CUTANEOUS MANIFESTATIONS
CUTANEOUS VASCULAR LESIONS
CUTANEOUS VASCULAR
LESIONS
ANGIOKERATOMA
Angiokeratomas, are the cutaneous hallmark of Fabry disease.11-13 Present in 70% of males and 39% of females,13,14 angiokeratomas are pinpoint to 4 mm diameter, dark-red to blue-black, macular and papular lesions that do not blanch on pressure (Fig. 127-3). Overlying hyperkeratosis is variable and frequently absent at sites other than the genitalia and umbilicus.
Natural history of Fabry disease
Cardiac disease
CNS disease
Renal disease
GI disease
Initial symptom
Corneal opacities
Angiokeratomas
Acroparesthesias
0
20 40+ Age (years)
Diagnosis Mean age for Classic Fabry: 50 years (age range: 40-56 years) Quality of life
21
A
B
Angiokeratomas appear between the ages of 5 and 15 years in males and the ages of 8 and 25 years in females.15-17 They were a presenting feature in 50 (14.2%) of 352 pediatric patients,18 in 114 (31.7%) of 359 adult males (n = 359), and in 13 (11.2%) of 118 adult females (n = 118).19 They may be widespread or grouped. In males, lesions are typically within the “bathing trunk” area (genitals, buttocks, lower abdomen, umbilicus, groins, inner thighs and sacrum). Angiokeratomas are also seen on the proximal limbs, particularly their medial aspects, the elbows and knees, the palms and soles, and over the distal phalanges of the digits (Fig. 127-4). Lesions may occur on the lips, particularly along the vermilion border, and occasionally on the mucosal surfaces (Fig. 127-5). They rarely occur elsewhere on the face. In females, angiokeratomas are usually sparsely distributed (Fig. 127-6), and may occasionally occur in a dermatomal distribution.13 The most common sites are the trunk and proximal limbs.13 Female genital lesions are relatively infrequent.
HISTOPATHOLOGY
HISTOPATHOLOGY
Histology typically shows dilated, ectatic capillaries in the papillary dermis, a variably thinned epidermis centrally, with epidermal acanthosis at the edges of the lesion, and variable degrees of overlying focal compact
2293
21
A
C
E
F
orthohyperkeratosis (Fig. 127-7A). Endothelial, perithelial, perineural, eccrine, smooth muscle cells, and fibroblasts are filled with cytoplasmic vacuoles containing glycosphingolipid that can be visualized with toluidine blue stains.20 Characteristic, electron-dense, lamellated,
2294
B
D
intracytoplasmic vacuolar inclusions (Zebra bodies) are typically seen on electron microscopy (Fig. 127-7B-D).21 They exhibit a pattern of alternating light and dark 4- to 6-nm bands. These inclusions may be present in biopsies of angiokeratoma or from normal skin. They may
A
21
B
be absent in the skin of heterozygous females. Electron microscopy and immunoelectron microscopy using anti-Gb3 antibodies are useful tools when other diagnostic tests are inconclusive.22
ANGIOMAS AND TELANGIECTASES
Up to a third of Fabry males and two-thirds of Fabry females do not have angiokeratomas.16,17 A proportion have no cutaneous vascular lesions,1 others have 1- to 2-mm diameter bright red macular angiomatous lesions, which could represent early angiokeratomas or macular hemangiomas clinically.11 Patients with widespread papular angiomas, clinically and histologically in keeping with cherry angiomas, are also recognized. In one series, patients with a predominantly cardiac phenotype (N215S mutation) rarely had classical angiokeratoma, but a third of males in this group had widespread (>100)
cherry angiomas.23 Because the prevalence of cherry angiomas in the general population may be as high as 50%,18 their significance in patients with Fabry disease is not clear. Telangiectases are also common,13 and may be distinguished from angiokeratomas and macular and cherry angiomas by the presence of blanching on diascopic pressure. Registry data show that they are more common in males (23%) than females (9%).13,14 Although based on patient recall, data suggest that telangiectases appear later than angiokeratomas, with a mean age at onset of 26 years in males (SD 17, range: 3-70 years) and 42 years in females (SD 22, range: 5-73 years).13 In a majority of cases, telangiectases occur at sun-exposed sites such as the face and V of the neck, often in patients with skin Type I or Type II. Occasionally, telangiectases may be seen at unusual sites, such as the flanks, groin, and elbow and knee flexures. Dermoscopy of these areas reveals dilated, tortuous upper dermal vessels (see Fig. 127-3). This tends to occur in patients with widespread angiokeratoma and full phenotypic expression of disease.
RELATIONSHIP TO DISEASE SEVERITY
In both sexes, the presence of cutaneous vascular lesions, namely telangiectases and/or angiokeratomas, is associated with higher disease severity scores and a higher prevalence of major organ involvement.13,14 Thus, cerebrovascular involvement (stroke and transient ischemic attack) was present in 38% of females and 32% of males with cutaneous vascular lesions, but in only 12% of females and 9% of males without. Cardiac involvement occurred in 80% of females and 73% of males with cutaneous vascular lesions versus 38% of females and 49% of males without; renal involvement occurred in 62% of females and 72% of males with cutaneous vascular lesions versus 29% of females and 42% of males without. Similar differences are observed for hypertension, eye, ear, GI, and other neurologic involvement.14 These findings demonstrate the importance of dermatologic assessment and its possible predictive value in terms of systemic morbidity.
2295
21
A
C
B
D
FACIAL FEATURES
FACIAL FEATURES
A “pseudoacromegalic” facial appearance has been described in some families with Fabry disease (Fig. 127-8). This feature is documented in 2 larger studies. An assessment of 38 patients by a panel of 3 clinical geneticists was based on standardized medical photography24 and identified (in order of decreasing frequency) periorbital fullness, prominent ear lobes, bushy eyebrows, recessed forehead, pronounced nasal angle, generous nose/ bulbous nasal tip, prominent supraorbital ridges, shallow midface, full lips, prominent nasal bridge, broad alar base, coarse features, posteriorly rotated ears, and prognathism. A second study, in which facial dysmorphism was objectively assessed by three-dimensional dense surface modeling and
2296
anthropometric analysis using a three-dimensional photogrammetric camera, facial features in 20 males and 22 females with Fabry disease were compared to controls.25 This study confirmed, in males, and less prominently in females, the presence of the facial features noted above. The presence of these facial features should prompt appropriate investigations for Fabry disease. The Fabry face appears more common in patients with widespread angiokeratomas and classical disease at the more-severe end of the spectrum. Moderate or marked facial changes were present in 26 (63%) of 41 males with a classical disease phenotype; 16 (61%) of the 26 males had angiokeratoma corporis diffusum. In contrast, facial features were present to a very mild degree (periorbital puffiness only) in 2 (10.5%) of 19 males with a predominantly cardiac phenotype of disease.14
LOWER-LIMB EDEMA AND LYMPHEDEMA
LOWER-LIMB EDEMA AND
LYMPHEDEMA
Edema and lymphedema, particularly affecting the limbs, is frequently observed in Fabry patients (Fig. 127-9). Lymphedema was cited in the original description of Fabry disease and is
21
recognized in other lysosomal storage diseases such as α-N-acetylgalactosaminidase deficiency.26 Case reports document it as an unusual presenting feature of the condition,27 and some suggest that familial lymphedema and Fabry disease might be linked.28 Registry data from the Fabry Outcome Survey on more than 700 patients confirm that reversible peripheral edema of the lower limbs was present in 25% of Fabry males and 17% of Fabry females; lymphedema was present in 16% of Fabry males and 7% of Fabry females. The mean age of onset was 37 years for males (range: 13-70 years) and a decade or so later for females.13 This is significantly more than the documented UK community prevalence of 1.33 per 1000 population.29 In the community it is 5 times more common in women. The mechanism underlying the changes is unclear, and the presence of the changes does not correlate with renal or cardiac involvement. Contributory factors may include glycosphingolipid accumulation,30 recurrent edema, and primary abnormality of the lymphatics.31,32
Using the technique of fluorescence microlymphography and measurement of lymph capillary pressure, Amann-Vesti et al demonstrated fragmentation of the microlymphatic network in 5 of 5 hemizygous males and 5 of 5 heterozygous females but in none of 12 healthy controls. Severe structural and functional changes in the initial lymphatics of the skin were present, even when lymphedema was not manifest.32 Significantly higher serum levels of vascular endothelial growth factor (VEGF)-A, a glycoprotein known to stimulate endothelial cell migration and proliferation, and to increase microvascular permeability and vasodilation, have been identified in Fabry patients.33
The possibility that abnormalities in expression or function of recently identified mediators of lymphangiogenesis (VEGF-C/-D and VEGF-Rs)34 may be involved in the development of lymphedema in Fabry disease remains to be investigated.
ABNORMALITIES OF SWEATING
ABNORMALITIES OF
SWEATING
Reduced sweating is a classical feature of Fabry disease and thought largely to be a consequence of autonomic neuropathy, although substrate accumulation within sweat glands may play a role.35,36 Hypohidrosis was reported by 53% of males and 28% of females, with earlier onset in males (mean age: 23 vs 26 years). Anhidrosis was described by 25% of males, but only 4% of females.8 Heat intolerance is a commonly associated and disabling symptom, resulting in reduced exercise tolerance, nausea, dyspnea, light-headedness, and headache, or complete collapse with loss of consciousness. Previous studies36,37 demonstrated an improvement in sweating in Fabry patients undergoing enzyme replacement therapy (ERT). Hyperhidrosis may also occur, more often in females than males: 44 (11.9%) of 369 females versus 22 (6.4%) of 345 males.38 This is higher than the
2297
21
estimated prevalence of 1.0% to 2.8% of the general population in the United States.39 In a majority of Fabry patients, hyperhidrosis affects palms and soles and is not generalized.38
RAYNAUD PHENOMENON
RAYNAUD PHENOMENON
“Cold intolerance” and the development of pain in the extremities in cold environments is a frequent complaint among Fabry patients. Registry data has documented Raynaud phenomenon in 57 (8%) of 710 females and 71 (11%) of 644 males (data from Fabry Outcome Survey).14 Two newer studies identified Raynaud phenomenon in 15.3% to 38% of Fabry patients.40,41 This is significantly higher than the prevalence of 3% to 22% reported in the general population, in which males are less commonly affected (0.5% to 16%) than females (2.5% to 22%).41 The reversal of the sex ratio and high prevalence in males suggest a possible causal link to the underlying Fabry disease. In a fluoroscopic nailfold capillaroscopy study42 that examined 25 Fabry patients (17 males), significantly more bushy capillaries and clusters were identified in cases (72%) than controls (10%). Morphologic and functional abnormalities of nailfold capillaries were present. In a further capillaroscopic study of 32 Fabry patients, 25 (78%) of whom were on ERT, the only significant difference identified was an increase in ramification of capillaries.41 Although the abnormal vasoreactivity of digital vessels could be related to autonomic dysfunction, abnormalities of nitric oxide synthetase, and increased oxidative stress in vascular endothelium and smooth muscle, might also be important triggers.43
NONCUTANEOUS FINDINGS
The noncutaneous findings in Fabry disease are illustrated in Figure 127-2 and listed in Table 127-1 in relation to their typical age of onset. Fabry disease is a progressive, multisystem disorder that typically results in a global reduction in quality of life for affected individuals. Symptoms in childhood typically relate to lethargy, tiredness, pain, cutaneous abnormalities, changes to sensory organs, and, often, GI disturbances. In early adulthood, patients may suffer extension of any of the above symptoms and often develop lymphedema, proteinuria and the first signs of renal, cardiac, or CNS/cerebrovascular disease. In late adulthood (age >30 years), symptoms include worsening of the above and more-severe organ dysfunction (cardiac disease, renal disease, and cerebrovascular disease).
NEUROLOGIC FINDINGS
NEUROLOGIC FINDINGS
Acroparesthesias occur in 80% to 90% of affected individuals and typically occur in the first decade.16
2298
■Renal
■Renal
■Chronic renal failure
■Chronic renal failure
■Cardiovascular
■Cardiovascular
■Hypertension
■Hypertension
■Coronary vascular disease
■Coronary vascular disease
■Left ventricle hypertrophy
■Left ventricle hypertrophy
■Valve disease
■Valve disease
■Arrhythmias
■Arrhythmias
■Elevated high-density lipoprotein
■Elevated high-density lipoprotein
■Neurologic/psychiatric
■Neurologic/psychiatric
■Acroparesthesias
■Acroparesthesias
■Cerebrovascular disease
■Cerebrovascular disease
■Dementia
■Dementia
■Depression
■Depression
■Increased suicide ideation
■Increased suicide ideation
■Ocular
■Ocular
■Cornea verticillata
■Cornea verticillata
■Conjunctival vessel aneurysms
■Conjunctival vessel aneurysms
■Increased tortuosity of retinal vessels
■Increased tortuosity of retinal vessels
■Fabry cataract
■Fabry cataract
■GI
■GI
■Diarrhea
■Diarrhea
■Nausea
■Nausea
■Vomiting
■Vomiting
■Postprandial flank pain
■Postprandial flank pain
■Malabsorption
■Malabsorption
■Others
■Others
■Hearing loss and tinnitus
■Hearing loss and tinnitus
■Hypothyroidism
■Hypothyroidism
■Obstructive airway disease
■Obstructive airway disease
■Osteopenia and osteoporosis
■Osteopenia and osteoporosis
■Anemia
■Anemia
Patients describe the sensation as pain, feeling like pins and needles in hands and feet often radiating proximally. Triggers include increased body temperature, exercise, and stress. Pain typically declines over time, which may be the result of damage to nerve fibers as a result of inflammation and substrate accumulation.
SENSORY ORGAN ABNORMALITIES
SENSORY ORGAN
ABNORMALITIES
The most common ocular finding is cornea verticillata (opacities in the cornea characterized by 1 or more lines radiating from near the center of the cornea), which occurs in more than 90% of Fabry males and 70% of Fabry males. Other changes include increased tortuosity of retinal vessels, optic atrophy, cataracts, and lenticular changes. The extent of ocular abnormalities correlates with the overall extent of the disease.44 Ocular changes also correlate with the genotype, and are more severe in subjects with nonsense mutations.45 Tinnitus and high-frequency sensorineural hearing loss are also common manifestations, occurring in more than 50% of patients.16
Sudden deafness and dizziness resulting from vestibular pathology can also occur.16
GASTROINTESTINAL CHANGES
GASTROINTESTINAL
CHANGES
Cramping abdominal pain, nausea, diarrhea and, occasionally, constipation are frequent, and are often the presenting symptom.46 The pathogenesis is probably related to neurologic abnormalities.
ORGAN DAMAGE IN FABRY DISEASE
ORGAN DAMAGE IN
FABRY DISEASE
RENAL MANIFESTATIONS
Renal manifestations are seen in more than 90% of males.16 Microalbuminuria and hyperfiltration are early features; proteinuria is typically seen in the third and fourth decades; and progressive decline in renal filtering capacity occurs. Females frequently show proteinuria, although progression to end-stage renal failure is less common. A renal variant of Fabry disease has been described in patients with decreased,1 but not absent, α-galactosidase A activity; these patients lack other characteristic manifestations.
CARDIAC MANIFESTATIONS
Cardiac manifestations are a constant feature and increasingly recognized as the major cause of death in both male and female patients.47 Substrate deposition can be demonstrated throughout the myocardium, valves, and conduction system, and is often accompanied by an inflammatory cell infiltrate. A common presentation is with left ventricular hypertrophy, but mitral valve prolapse, arrhythmia, and coronary artery disease can all be present. A cardiac variant1 has been described in patients with reduced, but not absent, α-galactosidase A activity, and presents later in life (often when patient is older than age 40 years) with predominant cardiac symptoms. MRI scanning has a valuable role in detecting early involvement and in demonstrating scarring, which is a feature of advanced disease.48
CEREBROVASCULAR MANIFESTATIONS
Cerebrovascular manifestations include ischemic or hemorrhagic strokes occurring early in life, and transient ischemic attack and strokes affecting the posterior circulation. Stroke is often reported as the presenting feature of Fabry disease and Fabry disease should be considered in the differential diagnosis of
21
young patients with cryptogenic stroke.49 There is an increased prevalence of factor V Leiden among Fabry patients who have suffered stroke.50 Cognitive impairment can be demonstrated among older males and females with Fabry disease.51
OTHER MANIFESTATIONS
Global manifestations of Fabry disease include lethargy, tiredness, failure to thrive in children, and anemia. Depression is frequent and often underdiagnosed, affecting up to 50% of Fabry males and females.52 Sexual activity is often affected by the presence of angiokeratomas in the genital region and can lead to decreased self-esteem and libido. Autonomic dysfunction is underrecognized and can lead to hyperhidrosis, abnormal tear and saliva formation, abnormal cardiac reactivity, GI dysmobility, altered pain and temperature perception, and peripheral edema. Endocrine abnormalities are uncommon but osteopenia and hypothyroidism53 are well described. Pulmonary abnormalities are underrecognized; Fabry disease can lead to an obstructive airways pattern54 in some patients, but asthma is the presenting feature in other patients. In contrast to other lysosomal storage disorders, cognitive impairment is not generally seen; however, older patients (older than age 50 years) are increasingly seen with memory loss, global intellectual deterioration, and personality change.51
DIAGNOSIS
The definitive diagnosis of Fabry disease is usually delayed, with a mean time between the onset of symptoms and diagnosis of 15 years.55 The diagnosis must be confirmed by the demonstration of deficient α-galactosidase A activity in plasma, serum, or leukocytes and the identification of the pathogenic mutation. Female patients have variable, and, occasionally, even normal levels of enzyme activity, making DNA confirmation of the diagnosis essential. Biopsy of tissues, such as skin and kidney, demonstrates the presence of lipid deposition and multilamellated myelin bodies in electron micrographs (see Fig. 127-7). As Gb3 deposition starts in utero, prenatal diagnosis can be performed from chorionic villi or culture of amniotic cells, where low α-galactosidase A activity can be demonstrated.1 There is an increasing recognition of the importance of late-onset mutations; and it is very important that the pathogenicity of the mutation should be confirmed. Measurement of globotriaosylsphingosine can add valuable information.56
DIFFERENTIAL DIAGNOSIS
The pain associated with acroparesthesia is often misdiagnosed as rheumatoid arthritis, rheumatic fever, erythromelalgia, Raynaud disease, or simply as “growing pains.”16
2299
21
LOCALIZED FORMS ANGIOKERATOMA CORPORIS DIFFUSUM
Angiokeratoma of Fordyce Fucosidosis
Angiokeratoma of the vulva Aspartylglycosaminuria
Angiokeratoma of Mibelli Galactosialidosis
Solitary papular angiokeratoma Schindler/Kanzaki disease
Angiokeratoma circumscriptum β-Mannosidosis GM1-gangliosidosis/
Angiokeratoma circumscriptum β-Mannosidosis GM1-gangliosidosis/ β-galactosidase Sialidosis Idiopathic
β-galactosidase Sialidosis Idiopathic
The clinical diagnosis of angiokeratomas in Fabry disease may be difficult. Careful inspection of the skin may be required to distinguish them from purpura, petechiae, and angioma serpiginosum. They should also be distinguished from the solitary and localized forms of angiokeratoma that occur in the absence of underlying systemic disease (Tables 127-2 and 127-3).57
Widespread angiokeratomas occur in other lysosomal storage disorders which should be considered in the differential diagnosis. They include fucosidosis,58 where more than 50% of patients have the changes, α-N-galactosaminidase deficiency and galactosialidosis (47% to 50%) (see Tables 127-2 and 127-4). In addition to the distinct enzyme deficiency in each of these storage diseases and other clinical features, angiokeratomas from Fabry disease can be
differentiated by electron microscopic examination by the characteristic electron-dense, lamellar (zebra-like) inclusions within endothelial and other cell types.59-62 Chloroquine therapy may result in storage of biochemically and ultrastructurally similar inclusions in many of the same cells as Fabry disease and may result in similar clinical manifestations, including the development of angiokeratoma.63 Widespread angiokeratomas have also been described in association with tuberous sclerosis,64 juvenile dermatomyositis,65 and without an associated metabolic disorder.66
PROGNOSIS AND CLINICAL COURSE
Earlier studies suggested that males with Fabry disease typically die in the fourth or fifth decade, and females live perhaps 15 years longer. More recently, the life expectancy for males has been estimated as 58.2 years (compared to 74.7 in the general U.S. population) and 75.4 years for females (compared to 80 years in the general U.S. population).67 There is also evidence that, whereas renal failure was previously the most common cause of death, cardiac disease and cerebrovascular disease are increasingly common. Some of these changes may be a result of the impact of ERT.67,68
TREATMENT
TYPE AGE AT ONSET (YR) SEX CLINICAL ASPECT BODY SITE INHERITED ASSOCIATION
Fabry 5-12 Both, males > females Multiple, clustered dark-red to blue-black macules and papules; larger lesions warty (1-4 mm)
Any part of the body, especially bathing trunk area, umbilicus, lips
Yes, X-linked Acroparesthesias, heart and renal failure, stroke, cornea verticillata, deafness, etc.
Mibelli 10-15 Both, female > males Grouped, warty, dark-red papules (1-5 mm) Lateral aspect and dorsa of fingers and toes, hands, and feet
Yes, autosomal dominant Acrocyanosis and chilblains
Fordyce >60 Male Multiple warty, blue-black papules (1-4 mm) Genitals, mainly scrotum No Local venous hypertension
Vulva >60 Female Grouped, warty, blue-black papules (1-4 mm) Vulva No Local venous hypertension
Solitary papular 10-40 Both Single dark-red to black keratotic papules (2-10 mm) often bleed/thrombose
Circumscriptum Usually early
Both Unilateral plaque keratotic
Circumscriptum Usually early onset or at birth
Both Unilateral plaque keratotic dark-red papules, may bleed
onset or at birth
dark-red papules, may bleed
2300
Any part of the body, especially in lower extremities
No —
No Cobb syndrome; vascular malformations
Lower legs or
No Cobb syndrome;
Lower legs or foot may be zosteriform53
foot may be zosteriform53
vascular malformations
21
GROUP NAME ENZYME DEFICIENCY GENE
ELECTRON MICROSCOPY FINDINGS55 CLINICAL FINDINGS DERMATOLOGIC FINDINGS
Sphingolipidoses Fabry (OMIM #301500) α-Galactosidase A Xq22 Electron-dense lysosomal deposits
Acroparesthesias, heart and renal failure, stroke, cornea verticillata
Angiokeratoma corporis diffusum, telangiectasias, hypohidrosis/anhidrosis, lymphedema
β-Galactosidase 3p21-3pter Electron-lucent lysosomal dilation
GM1-gangliosidosis (OMIM #230500)
Facial dysmorphism, hematologic signs, mental retardation, organomegaly
Angiokeratoma corporis diffusum
Aspartylglycosaminidase 4q32-33 Electron-lucent lysosomal dilation
Glycoproteinoses Aspartylglucosaminuria (OMIM #208400)
Coarse facies, macroglossia, organomegaly, ocular findings, cardiac valve involvement
Angiokeratoma corporis diffusum, facial angiofi- bromas, oral fibromatosis, and leukokeratosis
α-Fucosidase 1p34 Electron-lucent lysosomal dilation
Fucosidosis (OMIM #230000)
β-Mannosidosis (OMIM #248510)
Mental retardation, coarse facies, growth retardation, recurrent respiratory tract infections, dysostosis multiplex, visceromegaly
Angiokeratoma corporis diffusum, widespread telangiectasias, acrocyanosis, purple transverse distal nail bands, increased vasculature in hands and feet, sweating abnormalities
β-Mannosidase 4q22-q25 Electron-lucent lysosomal dilation
Mental retardation, neuropathy, hearing loss, recurrent infections
Angiokeratoma corporis diffusum in bathing trunk area
Sialidosis II (OMIM #256550) Neuraminidase 6p21.3 Electron-lucent lysosomal dilation
Mental retardation, dysostosis multiplex, vacuolated lymphocytes, subtle coarse facial features
Angiokeratoma corporis diffusum
β-Galactosidase and neuraminidase 20q13.1 Electron-lucent lysosomal dilation
Multiple enzyme deficiency Galactosialidosis (OMIM #256540)
Dwarfism, gargoyle facies, mental retardation, seizures, corneal clouding, dysostosis multiplex, and hearing loss
Angiokeratoma corporis diffusum scattered along entire body, especially knees, elbows, and bathing trunk area
Kanzaki (OMIM #104170) α-Nacetylgalactosaminidase 22q11 Electron-lucent lysosomal dilation
OMIM, Online Mendelian Inheritance in Man.
Mental retardation, coarse facial features, ocular signs, hearing loss, neuropathy
Angiokeratoma corporis diffusum of entire body, more dense on the bathing trunk area, axillae, and breasts Telangiectasias on lips and oral mucosa
p y
Telangiectasias on lips
and oral mucosa
2301
21
Symptomatic
Angiokeratomas
■Liquid nitrogen
■Electrocoagulation
■Surgical excision
■Laser (pulsed-dye 585 nm, neodynium YAG [Nd:YAG] 1064 nm, combined pulsed-dye and Nd:YAG) and intense pulsed light
Lymphedema
■Manual lymphatic drainage massage and compression
Hyperhidrosis
■Aluminium chloride hexahydrate
■Electrophoresis
■Botulinum toxin
■Glycopyrrolate sodium
Raynaud phenomenon
■Avoid smoking, cold and vasoconstrictor therapies
■Losartan
■Diltiazem
■Fluoxetine
■Sildenafil
Pain
■Avoid triggers
■Diphenylhydantoin
■Carbamazepine
■Gabapentin
Stroke
■Antiplatelet
■Anticoagulant
Hearing
■Hearing aid devices
■Avoid noise trauma
Lungs
■Avoid smoking
■Bronchodilators
GI
■Pancrelipase
■Metoclopramide
Cardiovascular
■Antihypertensive drugs
■Antiarrhythmia drugs
■Artificial pacemakers
■Implantable defibrillators
■Coronary bypass
Chronic renal failure
■Angiotensin converting enzyme inhibitors
■Hemodialysis
■Allograft transplant
Specific Enzyme replacement
Specific Enzyme replacement
■α-Galactosidase B (Fabrazyme)
■α-Galactosidase B (Fabrazyme)
■α-Galactosidase A (Replagal)
■α-Galactosidase A (Replagal)
■Migalastat (for patients with amenable mutations)
■Migalastat (for patients with amenable mutations)
The advent of ERT has meant that patients and families are increasingly assessed in a specialist center, where they will have access to a physician or pediatrician, dermatologist, and possibly cardiologist/
2302
nephrologist/neurologist. Access to audiologists, ophthalmologists, gastroenterologists, and psychiatrists/ counselors, as well as to genetic counselors and nurses is desirable and available at larger centers.
SYMPTOMATIC THERAPIES
SYMPTOMATIC THERAPIES
Individual end-organ manifestations of disease are treated symptomatically (see Table 127-5).
SKIN-DIRECTED THERAPIES
Although clearance of Gb3 from the skin following ERT is reported,69,70 this does not necessarily translate to clearance of angiokeratomas. The variety of vascular lesions seen in Fabry disease can, however, be treated effectively with quasicontinuous wave and pulsed lasers, and intense pulsed light systems.71,72 Newer lasers, which combine a high-powered pulsed-dye laser and a 1064-nm long-pulse Nd:YAG laser penetrate deeper and seem to work particularly well for the larger genital angiokeratomas.73,74
Prophylactic therapy with grade II graduated belowknee compression hosiery can prevent the development of lymphedema in patients whose edema is still fully reversible. Once lymphedema is established, control can be maintained in up to 80% of patients by regular skin care, exercise, manual lymphatic draining and/or selfmassage, and the use of appropriate specialist bandaging and hosiery for lymphedema.75
There is some evidence that hypohidrosis improves with ERT. For those patients with hyperhidrosis, treatment options include the use of topical aluminium chloride hexahydrate, tap water iontophoresis and local botulinum toxin injections, oral glycopyrrolate sodium up to 2 mg three times daily (a well-tolerated anticholinergic with minimal side effects, provided there are no cardiac contraindications) and chemical or endoscopic sympathectomy.76,77
For patients with cold extremities, standard measures, such as stopping smoking, avoiding vasoconstricting medications such as beta-blockers, and maintaining warm hands and feet with suitable clothing during winter months are generally recommended. Drug therapies with proven efficacy for Raynaud phenomenon in other patient groups include angiotensin II receptor antagonists, calcium channel blockers, fluoxetine, and sildenafil.78,79 Possible cardiac and renal contraindications should be taken into account before prescribing these drugs in Fabry patients.
THERAPIES DIRECTED AT OTHER ORGAN SYSTEMS
Pain is the most disturbing and early symptom, and can sometimes be partially managed with diphenylhydantoin, carbamazepine, or gabapentin. Patients
should be encouraged to identify, and try to avoid, their personal precipitating factors. For the primary or secondary prevention of stroke and other vascular pathologies, such as retinal artery occlusion, antiplatelet and anticoagulant therapy might be needed. Metoclopramide and pancrelipase are used to reduce GI symptoms. Hypertension must be controlled, as it significantly affects 3 of the most affected organs: the kidney, brain, and heart. Angiotensin-receptor blockade should be undertaken at the first sign of proteinuria and is an important adjunct to ERT in slowing the decline in renal function. Fabry patients are good candidates for kidney transplant in the event of endstage renal failure and the role of ERT in this situation is to preserve the function of other organs. Ancillary care from a cardiac perspective includes consideration of antiarrhythmics, artificial pacemakers, and surgery, including septal ablation and cardiac transplantation.
SPECIFIC THERAPEUTICS: ENZYME REPLACEMENT THERAPY
SPECIFIC THERAPEUTICS:
ENZYME REPLACEMENT
THERAPY
Two formulations of ERT have been developed: agalsidase beta (Genzyme) and agalsidase alpha (Replagal). Only agalsidase beta is licensed in the United States, whereas both formulations are available in most other parts of the world. Agalsidase beta is administered at a dose of 1 mg/kg biweekly and is manufactured using a recombinant technology in a Chinese hamster ovary cell line. Agalsidase alpha is given at a dose of 0.2 mg/kg biweekly and is manufactured using a gene activation methodology in a human fibroblast cell line. The efficacy of both ERT formulations has been demonstrated in randomized controlled trials,80-82 and both improve biochemical (eg, levels of Gb3 in plasma, urine, and tissue biopsy) and clinical parameters. The main clinical parameters chosen for study are renal function, pain, cardiac size and function, and quality of life.83 Long-term effectiveness (with data over 10 years) has been demonstrated in Registry studies of both preparations.84,85 Both preparations are considered safe and well tolerated. The main side effects are infusion related (fever, temperature) and both preparations can induce antibody formation. However, the impact of antibodies on clinical effectiveness has not been demonstrated. The 2 ERT formulations are generally considered to be of equivalent effectiveness when used at their licensed doses.86 The optimum time for commencement of treatment has not been established. ERT is recommended for all symptomatic males and at the first sign of organ dysfunction in females. Patients receiving ERT should be regularly monitored with serial measurements of pain, quality of life, and renal and cardiac function. Their data should be entered onto Registries wherever possible.87 ERT improves hearing and GI symptoms. Direct beneficial effects of ERT on
21
CNS abnormalities have not been demonstrated and enzymes cannot cross the blood–brain barrier.
NEW TREATMENTS
NEW TREATMENTS
Chaperone-based enzyme enhancement therapy consists of small molecules that rescue misfolded/ mistrafficked enzymes from the lysosomes and transport them to the endoplasmic reticulum. Chaperonebased therapy can be administered orally and has demonstrated activity in subjects with amenable mutations and residual activity.88 Migalastat has been approved by the regulatory authorities in both the United States and Europe. Substrate deprivation (inhibition of an early step in the synthesis of glycosphingolipids) and the infusion of galactose are other therapeutic options that are still in the research stage. Gene therapy and hematopoietic cell transplantation are still being developed.
GENETIC COUNSELING
GENETIC COUNSELING
All families that have a member with Fabry disease should have an opportunity to receive genetic counseling. Given the X-linked nature of Fabry disease, female patients have a 50% chance of transmitting the gene to both their sons and daughters, whereas male patients will have no affected sons and 100% of daughters affected. The genotype–phenotype variability should also be stressed, especially among patients with subtle manifestations.
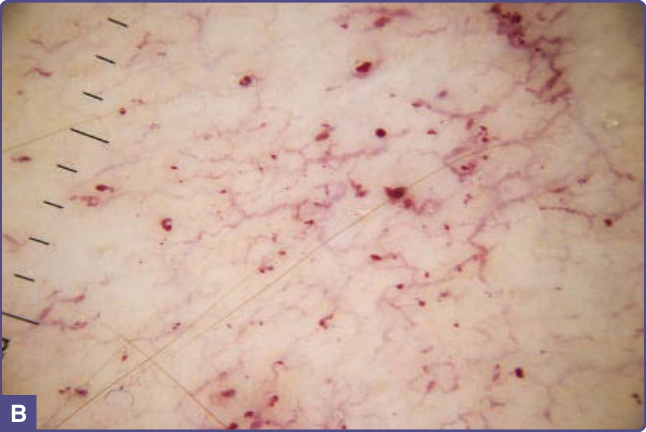
Figure 127-1 Metabolic pathway of globotriaosylceramide. α-Galactosidase A (α Gal A) metabolizes globotriaosylceramide to galactose (Gal) and lactosylceramide. Cer, ceramide; Glu, glucose.
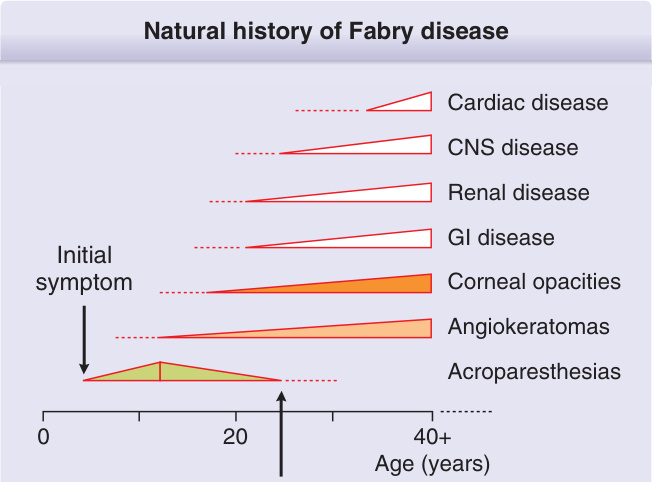
Figure 127-2 Natural history of Fabry disease. Acroparesthesias, angiokeratomas, corneal opacities, and GI symptoms are often the first manifestations. Diagnosis is usually delayed and occurs around the second decade.
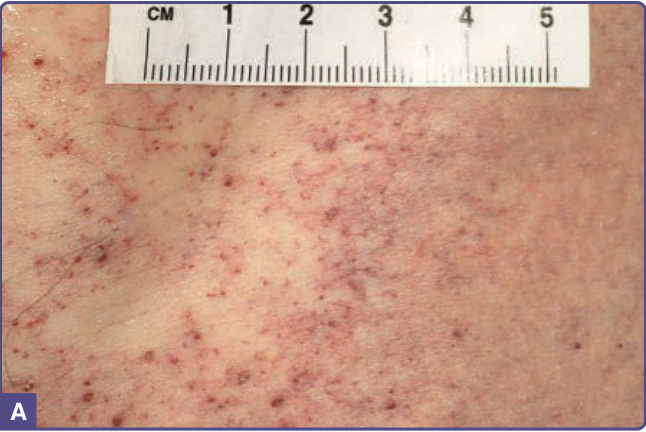
Figure 127-3 A, Angiokeratomas and telangiectatic vessels on the flank of a patient with angiokeratoma corporis diffusum. B, Dermatoscopic view showing angiokeratomas and upper dermal vessel tortuosity.
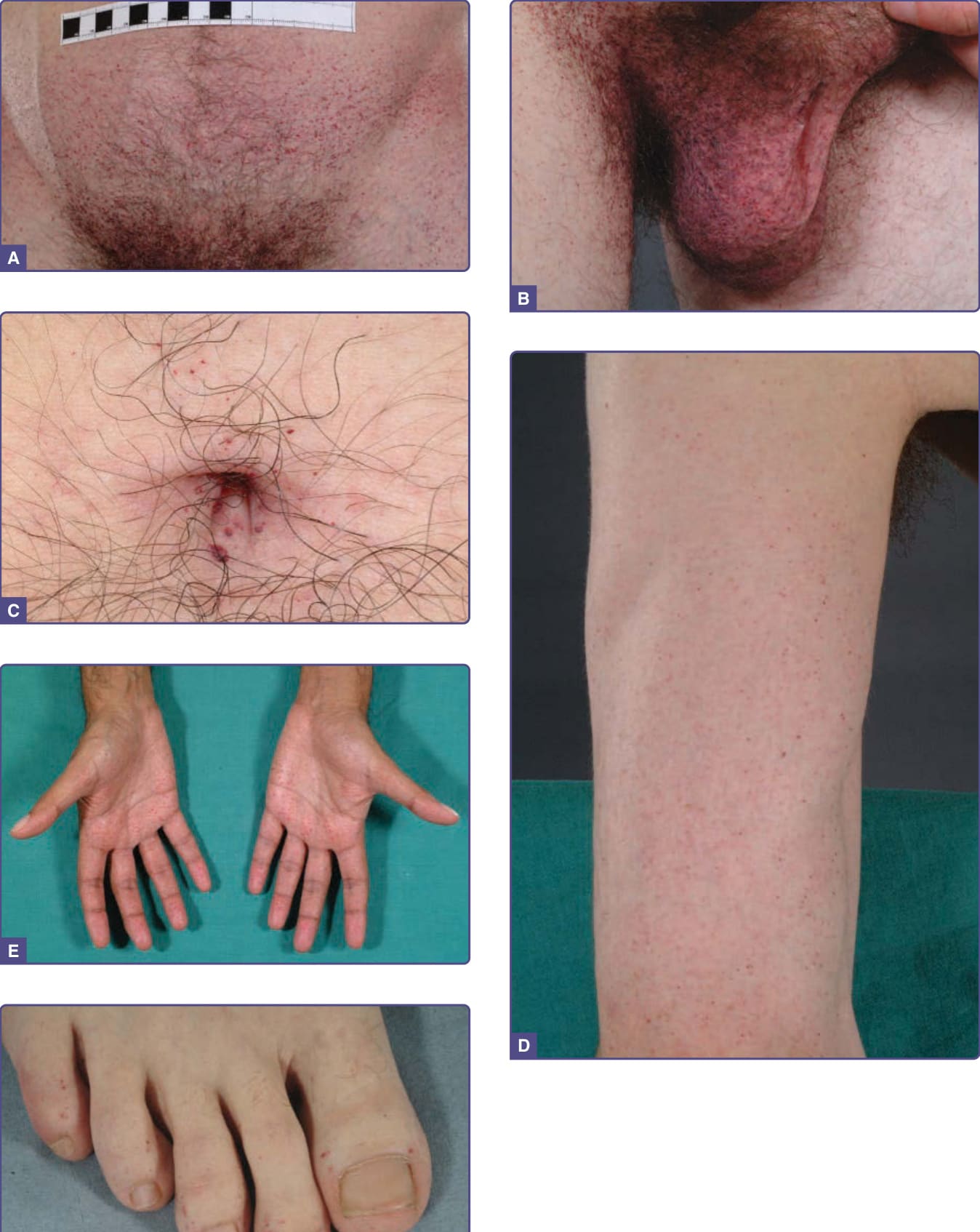
Figure 127-4 Distribution of angiokeratomas in Fabry males with complete phenotypic expression of disease. A, Lower abdomen; B, scrotum and medial thighs; C, umbilicus; D, upper limb; E, palms; F, toes.
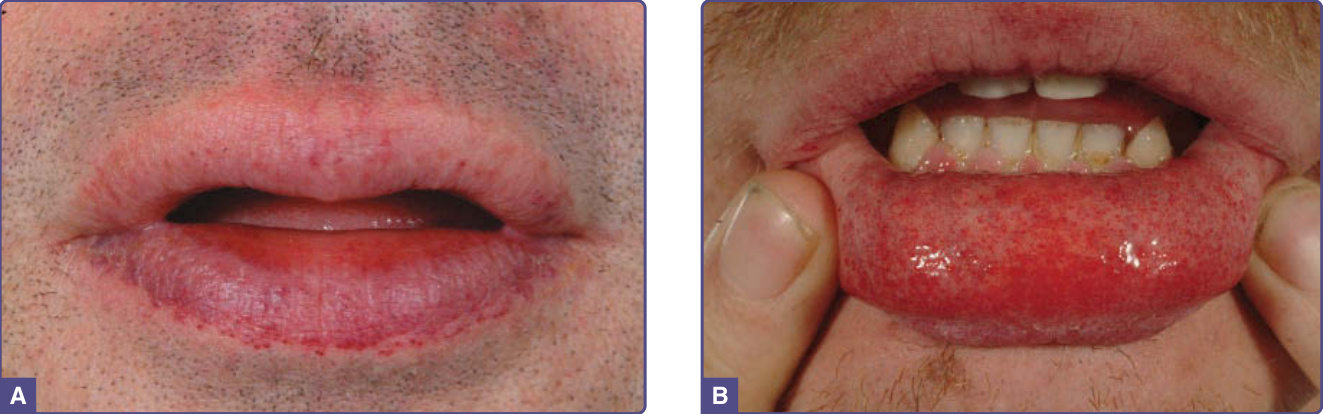
Figure 127-5 Angiomatous lesions on the lips. A, Concentrated along the vermilion border; B, on the mucosal surface of the lower lip.
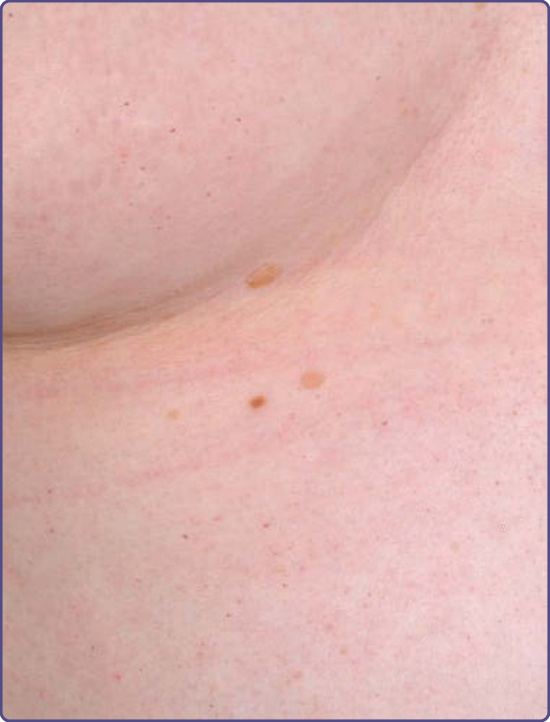
Figure 127-6 Sparsely distributed pinpoint dark-red macular and papular lesions on the trunk of a Fabry female.
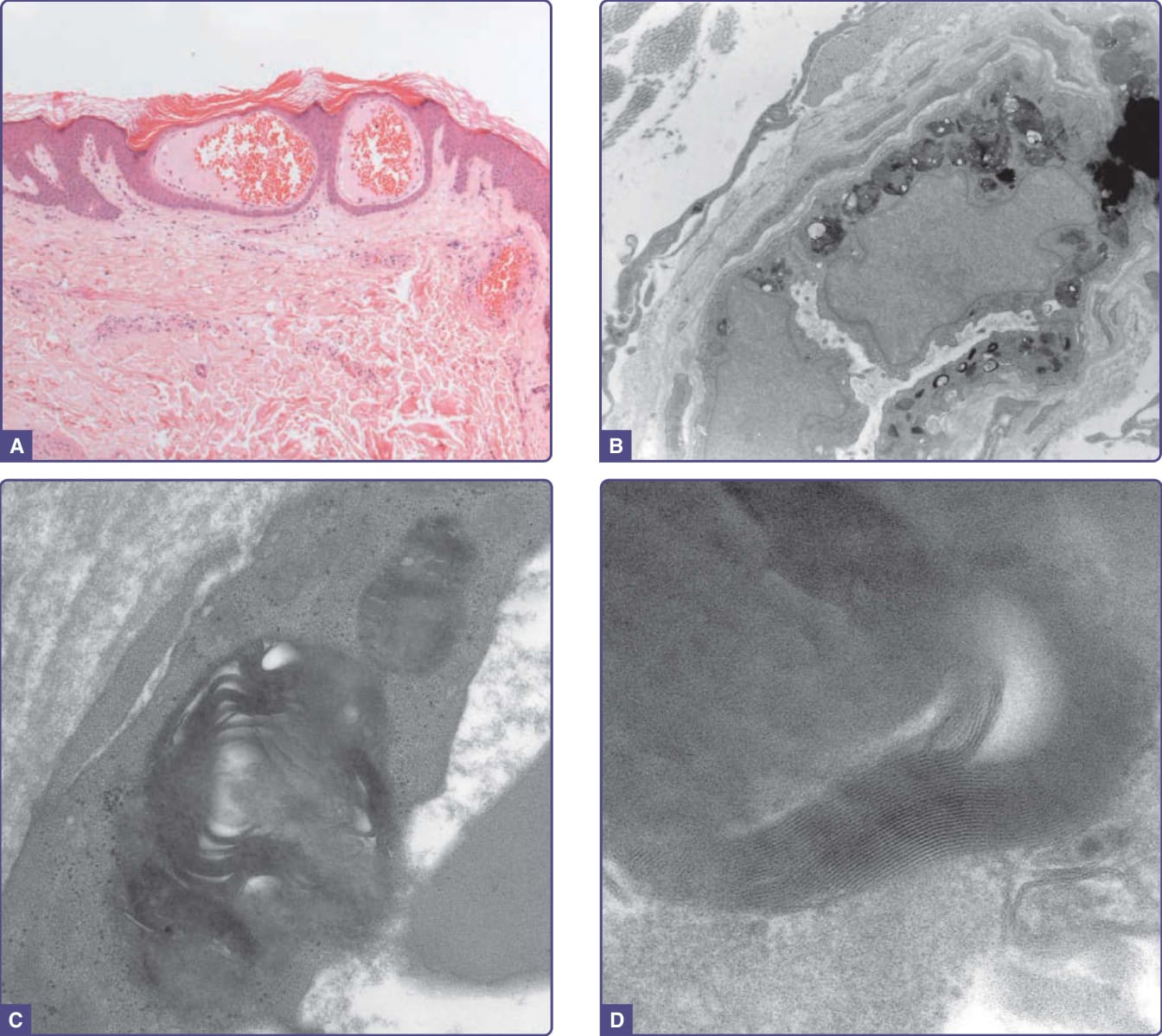
Figure 127-7 Histopathologic features of angiokeratoma. A, Light microscopy. Dilated capillaries in the papillary dermis with overlying epidermal thinning and mild orthohyperkeratosis. Acanthotic epidermis at the periphery. Hematoxylin and eosin (H&E) stain ×40 magnification. B-D, Electron microscopy. B, Multiple electron-dense inclusions in a cutaneous vascular endothelial cell (×9000 magnification). C, At higher power a lamellar or “zebra” body is seen (×63,000 magnification). D, At highest power (×223,000 magnification) light and dark bands with a periodicity of 4 to 6 nm are identified.
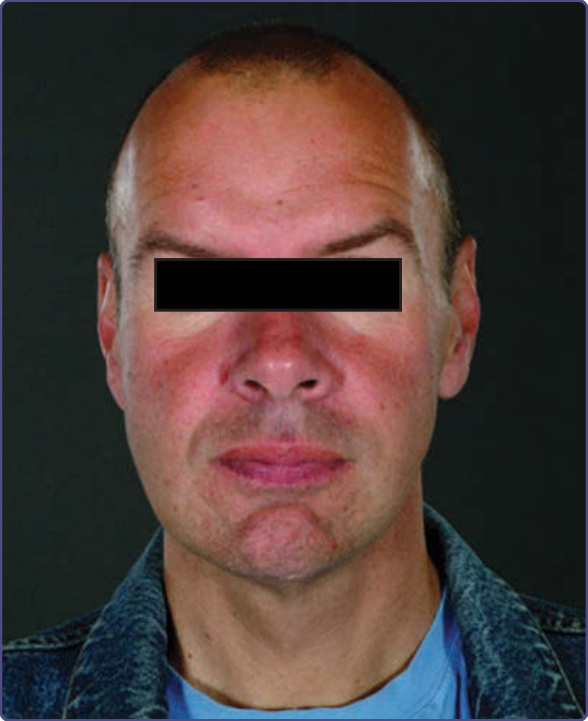
Figure 127-8 Typical facial features: prominent supraorbital ridges, periorbital fullness, large bitemporal width, bushy eyebrows, broad nasal base, full lips, and prominent chin.
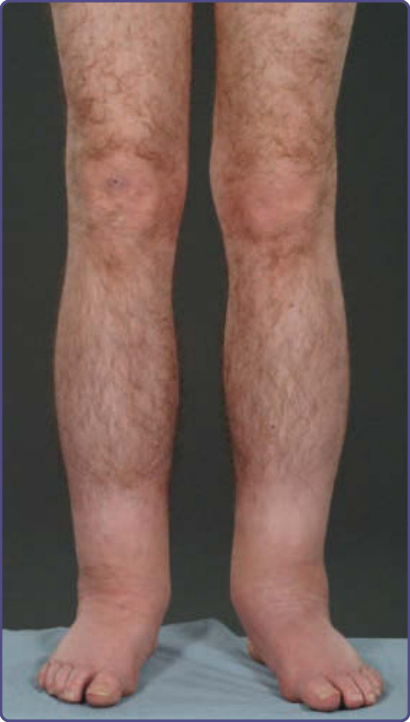
Figure 127-9 Bilateral lower-limb lymphedema in a 43-yearold Fabry male.

TABLE 127-1 Noncutaneous Findings

TABLE 127-2 Differential Diagnosis of Angiokeratomas
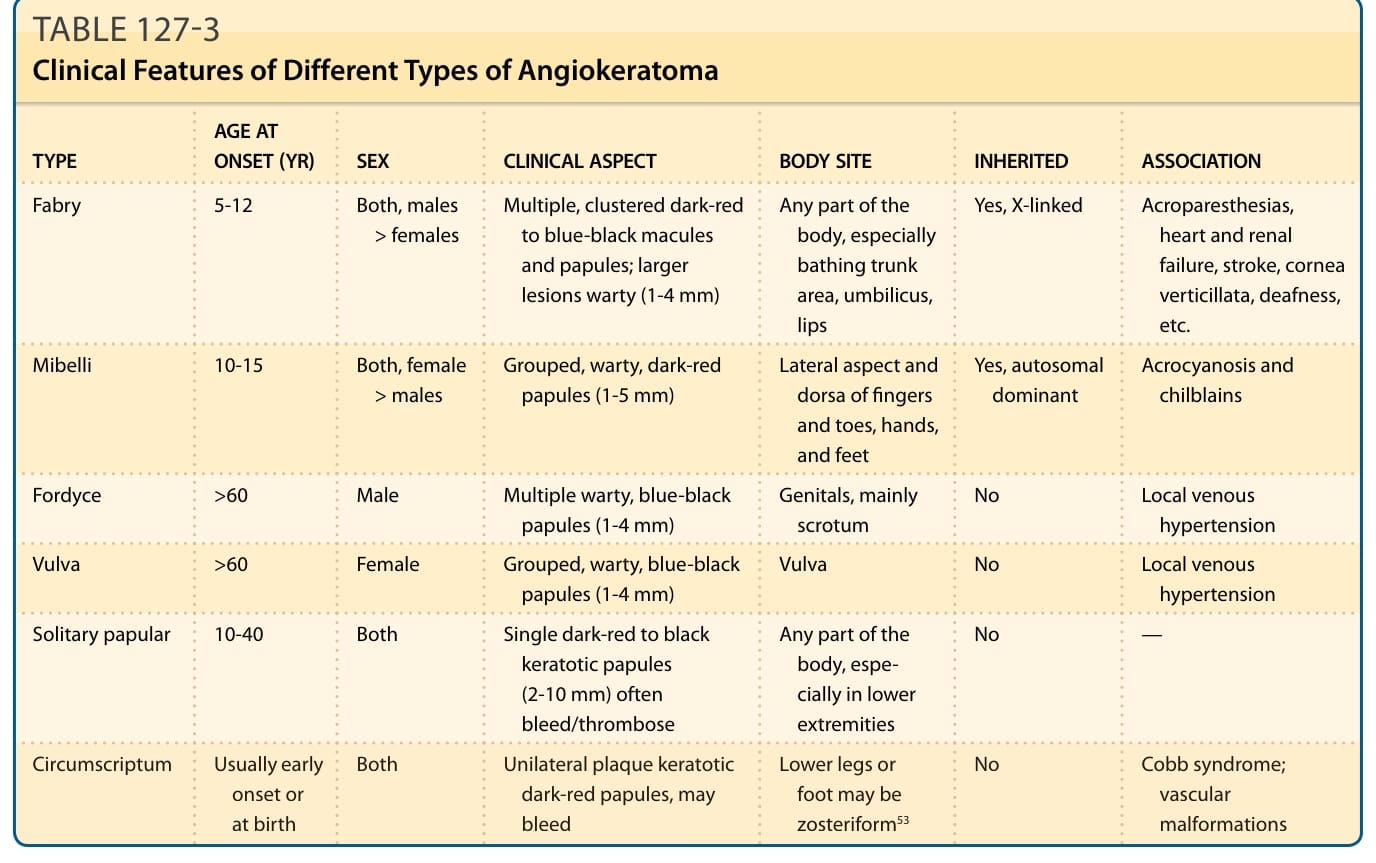
TABLE 127-3 Clinical Features of Different Types of Angiokeratoma
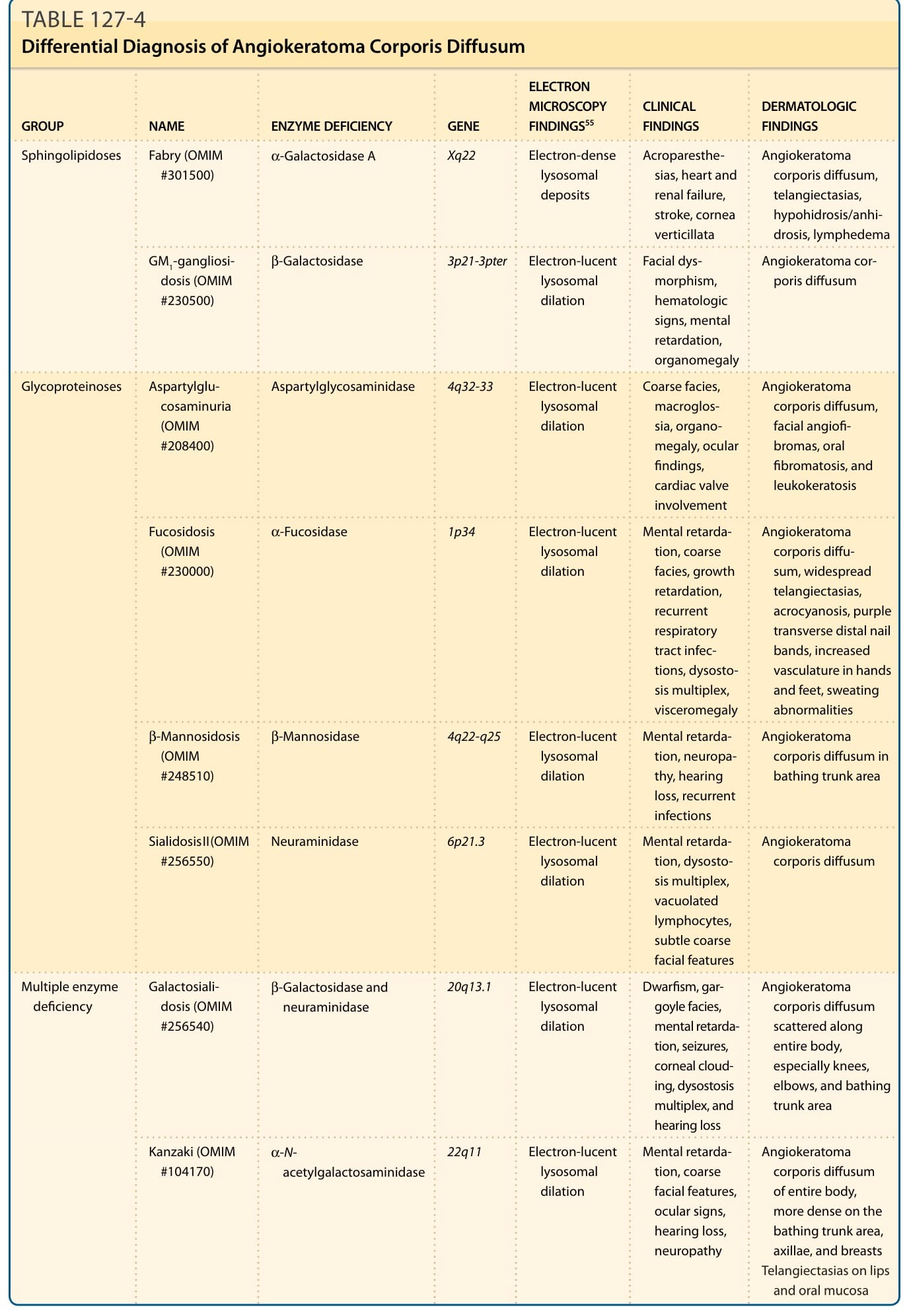
TABLE 127-4 Differential Diagnosis of Angiokeratoma Corporis Diffusum
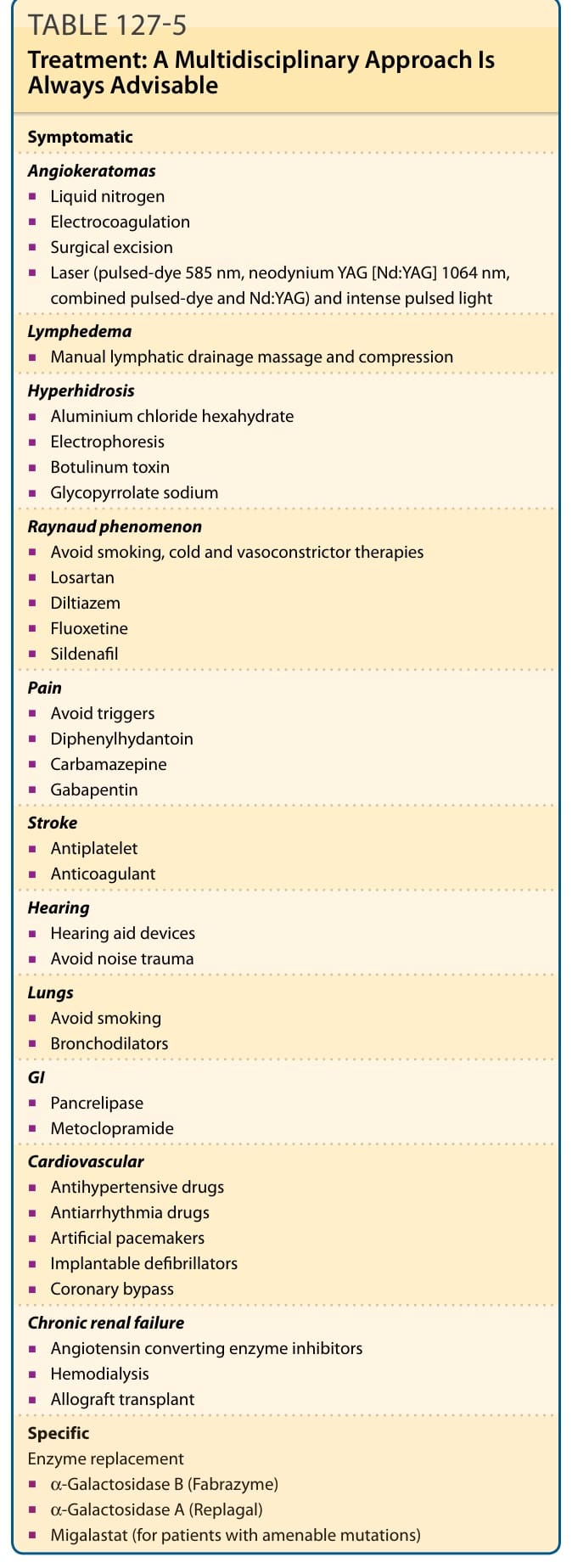
Table 127-5 outlines Fabry disease treatment. Fabry disease is a multisystem disorder and patients should be assessed within a multidisciplinary setting.