Cutaneous Vasculature
2
AT-A-GLANCE
■ The cutaneous vasculature is divided into a superficial, a deep, and a subcutaneous vascular plexus. The superficial vascular plexus is formed by parallel pairs of arterioles and venules connected via capillary loops that extend into the dermal papillae. These segments may individually or conjointly respond to exogenous or endogenous stimuli, thereby influencing skin disease expression.
■ Skin microvessels are formed from an endothelial cell lining that is supported by mural cells that are pericytes in most of the microvasculature but smooth muscle cells in the larger arterioles and venules and resident perivascular leukocytes, including T cells, macrophages, mast cells, and dendritic cells.
■ Skin microvessels, like other microvessels, perform three important constitutive functions: regulating fluidity of the blood, forming a barrier that separates and controls transfer of molecules and cells between circulating blood and tissue, and regulating local blood flow.
■ Control of blood flow through skin microvessels have a special and critical role in thermoregulation not performed by other segments of the vasculature.
■ Skin microvascular cell morphology and gene expression and function are altered by acute or chronic inflammatory skin diseases and in cancers. These processes may involve formation of new blood vessels (angiogenesis) or remodeling of preexisting vessels.
■ Skin-specific microvascular responses may be influenced by keratinocyte-derived and other environmental-derived factors.
INTRODUCTION
The blood vascular system is a continuous series of hollow tubes that are lined by a one-cell-thick layer of epithelium-like mesenchymal cells, the endothelium, and are supported by various mural cells, typically pericytes (PCs) in microvessels and smooth muscle cells (SMCs) in larger caliber vessels. All vascular endothelial cells (ECs) share common features and functions and hence may be collectively described as one cell type. However, ECs from one segment of the
vascular system may differ in significant ways from the ECs at other anatomic sites. Blood vessel ECs also differ from lymphatic ECs, which are not discussed in this chapter. Mural cells have distinct embryologic origins throughout the blood vasculature, but little is known about their variation with anatomic location. Consequently, these cells will be discussed as if they were homogeneous, but such descriptions should be regarded with caution.
STRUCTURE AND ORGANIZATION OF THE SKIN VASCULATURE
Approximately 3 decades ago, Irwin Braverman1
described the organization of the vascular network of human skin. The human dermal vasculature consists of two interconnected systems, a superficial and a deep vascular plexus (DVP) with additional vascular networks surrounding sweat glands and hair follicles (Fig. 9-1). The superficial vascular plexus (SVP) is composed of paired arterioles and venules that form an interconnected network of vessels coursing on a plane parallel to and just beneath the epidermal surface. Capillaries arise from the arterioles, extend upward within the papillary dermis at sites between the epidermal rete ridges, and then loop back down to the venules, forming arcade-like structures. Whereas the basement membrane of the arterioles appears homogeneous by electron microscopy, that of the venules is multilayered. In normal skin, most of the capillary loop is invested with a basement membrane that resembles that of the arteriole, acquiring a venule-like investiture only at the level of the deepest rete just proximal to the anastomosis with the venule of the SVP. There are no ultrastructural differences between capillary loops at different sites of the skin. The arterioles and venules of the SVP are connected by short, straight vessels to the arterioles and venules of a deeper planar network of anatomizing vessels, called the DVP (see Fig. 9-1). The plane of the DVP is parallel to that of the SVP and courses above the boundary between the reticular dermis and the underlying subcutis. The arterioles of the DVP are fed through penetrating vessels from the subcutis. The venules of DVP are drained by valve-containing veins that return to the subcutaneous fat. Capillary networks connecting the arterioles and venules of the DVP provide nourishment for the adnexal structures within the reticular dermis. The venules of the DVP serve as
Architecture of skin vasculature
Papillary dermis Epidermis
Vascular Segments:
Papillary loops
Reticular dermis
Superficial vascular plexus
Deep vascular plexus capillaries around hair and glands
Subcutis
Subcutaneous vascular plexus
Arteries and veins
portals of entry for leukocytes associated with inflammation of the adnexa or for inflammation within the reticular dermis itself. The walls of dermal arterioles are made of three layers: an intima composed of ECs, a media consisting of SMCs, and an adventitia containing some connective tissue-type cells. In the terminal arterioles, SMCs may be replaced by PCs that reside within the basement membrane of the ECs instead of in a distinct medial compartment. The walls of veins and venules have a similar structure, but compared with the corresponding arteries and arterioles, vessels of the venous circulation have a larger lumen and a thinner muscular wall and contain valves positioned at sites where the small vessels connect to larger ones. The walls of dermal capillaries and postcapillary venules consist of a monolayer of fenestrated ECs sitting on a simple basement membrane (Fig. 9-2). Capillaries are typically lined by a single file of highly curved ECs that enclose the lumen and are tightly connected to the adjacent ECs on either end, forming a tube with a lumen that is typically less than 10 μm in diameter. Postcapillary venules are of a somewhat larger caliber, are circumferentially lined by more than one EC, have looser connections between neighboring ECs, and have a multilayered basement membrane. The fenestrae of dermal capillary ECs are formed primarily by a protein called plasmalemmal vesicle protein (PV)-1 that was previously designated as Pathologische Anatomie Leiden-endothelial (PAL-E) antigen and used as a marker of microvascular ECs. PCs that reside within the basement membrane of dermal microvessels provide structural integrity to the walls of both capillaries and postcapillary venules and contribute to basement membrane synthesis, and when it occurs, remodeling. ECs outnumber PCs in skin microvessels, and each PC contacts multiple ECs and form adherens junctions.
2
Postcapillary venules of the SVP are also associated with various resident populations of leukocytic cells, including T cells, macrophages, dendritic cells, and mast cells. These cells, along with ECs and PCs, form the “perivascular extravasation unit”2 of the postcapillary venules, the segment of the vasculature through which circulating leukocytes can be recruited into the dermis. These perivascular resident leukocyte populations were identified more than 20 years ago and for the most part have not been reclassified as newer understandings of leukocyte lineages and developmental programs have been elucidated. For example, it is now appreciated that memory T cells can take up long-term residence in tissues, acquiring characteristics and functions distinct from those of circulating T-cell populations.3 The populations of perivascular T cells in noninflamed skin have not yet been categorized in comparison with circulating T cells or epidermal resident T cells with this new information in mind. Dendritic cells are now known to be quite heterogeneous with distinct developmental programs; each dendritic cell type serves different immunologic functions. Again, the types of perivascular dendritic cells have not been defined by these new criteria. Mast cells are also heterogeneous in terms of their granule contents and biosynthetic profiles, and it is unclear what human dermal perivascular mast cells may synthesize. Resident tissue macrophages are now known to originate either from more primitive embryonic origins, such as yolk sac or fetal liver, or from bone marrow– derived monocytes, and these populations also have distinct properties.4 Finally, dermal dendrocytes, also known as veil cells, form a resident population that envelop microvessels of the SVP, associate with T cells and mast cells, and express coagulation factor XIIIa and stabilin 1 in addition to other macrophage-like markers. There has been much debate as to whether these cells are bone marrow or mesenchyme derived, and with the new appreciation of resident macrophage heterogeneity, they may in fact be neither. In general, it is proposed that all such perivascular leukocyte cell types serve as sentinels for infection or injury, but it is possible that some of these populations also have constitutive vascular functions. Veins of the dermis form three kinds of anastomosing connections; large anastomoses of main trunks of the ascending veins, small anastomoses in the ascending sections of the small veins, and very small anastomoses from the small ascending veins reaching the papillary dermis. This anastomosing network of dermal veins is organized in polygons of various sizes, which is thought to play a role it outcomes of skin flap surgery.
PROPERTIES OF AND FUNCTIONS OF NORMAL SKIN VASCUALTURE5
117
See Table 9-1.
2
A
Schematic structure of capillaries
Pericyte
Endothelial cell
Basement membrane
Lumen
Tight junction
Weibel-Palade body Adherens junction
B
HEMOSTASIS
HEMOSTASIS
Blood is a fluid that is poised to become a gel at sites where vessel integrity is disrupted, beneficially forming a clot to limit extravasation but typically avoiding
118
formation of a thrombus within the vessel lumen that could occlude the vessel and limit perfusion. Vascular ECs are the principal cell responsible for maintaining blood fluidity, and they may be altered when needed to actively promote clot formation. Blood plasma contains the proteins of the coagulation system that
-
Hemostasis
-
Permselectivity
-
Perfusion
-
Thermoregulation
-
Immune surveillance
-
Hemostasis
-
Permselectivity
-
Perfusion
-
Thermoregulation
-
Immune surveillance
produce fibrin gels as well as platelets that can form a primary hemostatic plug. The EC lining prevents intravascular activation both of the coagulation system and of platelets yet keeps both systems poised to respond to injury. The key inhibitors of coagulation basally expressed by ECs are tissue factor pathway inhibitor (which prevents the dramatic increases of the enzymatic activity of factor VIIa on factors IX and X that is catalyzed by tissue factor), thrombomodulin (which redirects thrombin from cleaving and thereby converting fibrinogen to fibrin to target and activate protein C instead), and anticoagulant heparan sulfates (which activate antithrombin III to function as an inhibitor of thrombin and factor Xa). ECs also basally limit platelet activation by (1) producing
2
nitric oxide and prostacyclin (also called prostaglandin I2), which inhibits platelet activation by elevating intracellular cyclic guanosine monophosphate and cyclic adenosine monophosphate, respectively), (2) expressing ectoenzymes that degrade plateletactivating adenosine triphosphate and adenosine diphosphate to inert adenosine monophosphate, (3) minimizing the activation of thrombin (which activates platelets by cleaving protease activated receptor 1), and (4) masking basement membrane and interstitial collagens (which can be recognized by platelet surface proteins and serve as alternative activators of platelets) (Fig. 9-3).
PERMSELECTIVITY
PERMSELECTIVITY
Dermal ECs form a barrier that permits limited passage of fluid and solutes, allowing the blood to nourish the tissues while displaying “permselectivity” for macromolecules (Fig. 9-4). ECs also basally present a virtually impenetrable barrier for blood cells (except at specialized sites of high permeability and leukocyte trafficking). In general, arterioles and capillaries are less permeable to macromolecules because ECs
Anti-coagulant functions of endothelial cells
Inactivation of factors Va and VIIIa
Factor Xa
Thrombin
Protein S
Thrombin Fibrin
Activated protein C
Factors IX and X
Factors IXa and Xa
Platelets
VIIa + TFPI ATP ADP
Inactive AMP Fibrinogen
Prothrombin
Protein C
HS ATIII
Thrombin
TM
Basement membrane collagen
NO PGI2
ATPase or ADPase
TF
119
2
Permselectivity
Postcapillary venule y r a lli p a C e l o ir e tr A
Endothelium
Pericytes Shared basement membrane
H2O
Hydrostatic pressure
Smooth muscle cells
Tight junctions (impermanent macromolecules)
RBCs
H2O Oncotic pressure Adherens junctions (leaky to small molecules)
lining these vessels are connected to each other by tight junctions containing claudins (mostly claudin 5), junctional adhesion molecules (mostly JAM-A), and occludin (although occludin appears dispensable based on gene disruption in mice). Tight junctions prevent paracellular passage of macromolecules, limiting their crossing of the capillary EC lining to fenestrae or to vesicular transport, thereby enabling control of which molecules may pass (permselectivity). PCs may influence the barrier by stabilizing EC–EC contacts but are too sparse in skin microvessels to form a continuous surface that can directly block macromolecular transit. The capillaries, although narrower in diameter than other microvessels, have a cumulative surface area that vastly exceeds other parts of the microvasculature so that most transit takes place across this portion of the SVP. By allowing fluid and solutes to cross the EC lining by the paracellular route while preventing macromolecules to do so, dermal capillaries of the SVP allow hydrostatic pressure gradients near the arteriolar side of the capillary loop to drive fluid and solutes into the tissue and then draw them back into the bloodstream on the venular side of the capillary loop through oncotic gradients formed by the proteins retained within the vessel lumen. ECs of the postcapillary venules form few, if any, tight junctions and are instead held together primarily by adherens junctions, which are largely formed by VEcadherin and associated catenins. (Skin arteriolar and capillary ECs also form adherence junctions, but these seem dispensable once tight junctions form.) Venules are somewhat leaky to macromolecules under basal conditions, but macromolecular escape into the tissue is limited by the relatively small (in comparison with capillary) surface area of the venular EC lining. The
120
net result of these structural features is that dermal vascular ECs regulate rather than prevent molecular exchange between blood and tissues.
PERFUSION
PERFUSION
ECs regulate local blood flow primarily by acting on SMCs surrounding arterioles (Fig. 9-5). These are oriented almost parallel to the vessel’s circumferential or radial axis. ECs control SMC tone, which regulates the size of the vessel lumen and the resistance of the vessel to dilation. Vascular resistance along with blood pressure determines blood flow. PCs, which are also contractile, function largely to provide structural integrity to the terminal arterioles, capillaries, and postcapillary venules but may also contribute some to vascular resistance. Under normal circumstances, ECs synthesize both vasodilators, such as nitric oxide and prostacyclin, that reduce SMC tone and vasoconstrictors, such as endothelin, that increase tone. EC also express angiotensin-converting enzyme, which can generate a vasoconstrictor, angiotensin II, from angiotensin I, a circulating inert precursor. The balance between vasodilators and vasoconstrictors may be regulated by neural or hormonal signals. Importantly, inhibiting nitric oxide synthase-3 (also known as endothelial NOS) in ECs will cause vasoconstriction and increased resistance, suggesting that vasodilation is normally dominant. Recent evidence suggests that resident perivascular memory T cells specific for certain oxidized lipid adducts, can induce vasoconstriction through elaboration of cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-17A, although it is not known if this occurs in skin.6
2
Control of smooth muscle tone regulates perfusion
Endothelial cell
Receptors
ATI
ACE
NO PGI2 ET
Vasoconstriction Vasodilation
Smooth muscle cell
cGMP cAMP
Nerve
ATII
[Ca+2]
Norepinephrine and others
THERMOREGULATION
THERMOREGULATION
The skin is the source of thermal information and is an executive organ for thermal homeostasis. Temperature is sensed through primary somatosensory afferent nerves; skin blood flow is modified through sympathetic vasodilation or vasoconstriction. To conserve heat, cutaneous vessels constrict; for convective heat transfer from the core to the periphery, cutaneous vessels dilate, and blood flow increases. The skin blood flow is normally 250 to 300 mL/min but can vary from nearly zero in cold to as much as 6 to 8 L/min, which then covers approximately 60% of the cardiac output.7 Nonglabrous skin is subject to reflex thermoregulation by the sympathetic nervous system. The noradrenergic vasoconstrictor system is tonically active and is activated by cold exposure. Initial vasodilation to body heating is achieved by reflex removal of active vasoconstrictor tone. With further heating, the parasympathetic cholinergic nerves release predominantly acetylcholine, causing active vasodilation. Concomitantly, autonomic and sensory nerve fibers release neurotransmitters, such as substance P, calcitonin gene-related peptide, vasoactive intestinal peptide, and pituitary adenylate cyclase activating polypeptide, which directly or indirectly induce vasodilatation. Local factors such as venous congestions and increased transmural pressure also modulate skin blood flow. Glabrous skin contributes
to thermoregulation through shunts from the arterial directly into the venous beds, thereby bypassing capillary loops, the thermosensitivity of the hand being significantly greater than that of soles (Table 9-2). Dysregulation in the skin thermoregulation is seen in postmenopausal hot flushes related to changed estrogen levels. Estrogen has vasodilator function as
Nonglabrous Skin -Ambient temperature Basal activity of noradrenalic system -Cold: vasoconstriction Activation of noradrenalic system -Heat: vasodilation Shutdown of noradrenalic system -Further heating Parasympathetic cholinergic nerves
■Acetylcholine Autonomic and sensory nerve fibers
■Substance P
■Calcitonin gene-related peptide
■Vasoactive intestinal peptide
■Pituitary adenylate cyclase activating polypeptide
Glabrous Skin Additional thermoregulatory capacity through opening arteriovenous
Glabrous Skin Additional thermoregulatory capacity through opening arteriovenous shunts or closing arteriovenous shunt
121
shunts or closing arteriovenous shunt
2
demonstrated in estrogen receptor knockout mice. Estrogen replacement relieves hot flashes and reduces resting body temperature in these individuals. Individuals with Type 1 diabetes have greater skin blood flow relative to healthy control participants, potentially related to moderate vasodilation induced by a hyperinsulinemia and an impaired thermoregulatory response of skin vessels. The underlying mechanisms related to impaired heat responses remain largely unresolved. The absence of C-peptide produced in β cells or reduced nitric oxide bioavailability because of diminished NOS activity may play a role. A pathological thermoregulatory reflex is seen in Raynaud disease; patients have an exaggerated vasospastic response to cold or emotion, resulting in ischemia of the digits. Vasospastic attacks in Raynaud disease are restricted to cutaneous arteries supplying skin sites with a rich density of arteriovenous anastomosis, a situation at glabrous sites. These sites are normally targeted by sympathetic adrenergic vasoconstrictive activity in response to cold. Patients with Raynaud disease have increased sympathetic vasoconstrictive activity, which results in increased blood flow through arteriovenous anastomosis. Of note, bosentan, antagonizing endothelin A and endothelin B receptors, which has promising clinical effects in scleroderma, does not influence vasospasm in Raynaud disease. Patients with erythromelalgia have burning pain and vasodilation with erythema of the extremities. It is caused by mutations affecting the encoding SCN9A gene that leads to channelopathies. Although the primary cause of the abnormal vascular reaction is unknown, most patients have abnormal quantitative sudomotor axon reflex tests (measuring resting skin temperature, resting sweat output, and stimulated sweat output), some have abnormal adrenergic function, and others an abnormal cardiovagal function. Erythromelalgia most severely affects the palms and soles. It is therefore thought that arteriole–venule shunts are abnormally innervated or show an abnormal response. As pointed out earlier, these arteriole– venule shunts are mainly found in glabrous skin.
IMMUNE SURVEILLANCE
IMMUNE SURVEILLANCE
In addition of being a physical barrier, the skin has features of an immunologic barrier. Among resident cells housing in the skin are semiprofessional antigenpresenting cells such as keratinocytes, ECs, and macrophages and professional antigen-presenting cells such as Langerhans cells and dermal dendritic cells. In addition, the skin is populated by T cells. The majority of these cutaneous T cells are resident, largely within the epidermis, but as noted earlier, there also appear to be T cells resident in the perivascular compartment of the venules. In addition to resident populations, there are circulating T cells with a predilection for homing to skin. More than 30 years ago, the term skin-associated lymphoid tissues was created (subsequently renamed the skin immune system), which postulated the existence of
122
subsets of T lymphocytes displaying special affinity for the skin and that the acquisition of this affinity by T cells is determined signals received from resident cutaneous cells. After an immune response within the skin, a subset of memory T cells develops that can exit from the skin via the lymphatics to enter draining lymph nodes. These T cells may then reenter the skin upon rechallenge by the same anitigen,8 and they may retain a preference for skin sites. The first skin-associated protein expressed on a population of memory T lymphocytes was described and termed cutaneous lymphocyte antigen (CLA).9 CLA is a carbohydrate moiety on the surface of T cells closely related to sialylated Lewis X, which has homology to the core structure of P-selectin glycoprotein-1. CLA was shown to bind to E-selectin expressed on activated endothelium, but the latter is ubiquitously expressed on activated endothelium (albeit perhaps more transiently than in skin), suggesting that this is not the only mechanism regulating skin-specific leukocyte homing. A potential candidate providing additional specificity is the chemokine TARC (also called cutaneous T cell-attracting chemokine or CCL17) (Fig. 9-6), which is produced by cutaneous venules, induced in inflamed skin and absent on intestinal vessels.10 Its receptor, CCR4, is expressed on the CLA-positive memory T cells. The interaction of TARC and CCR4 triggers E-selectin–mediated adhesion of CLA positive T cells. However, skin infiltration in CCR4-/- mice is not impaired, indicating that further regulatory pathways exist. Another candidate for skin-specific recruitment of CLAhi+ T cells is the chemokine CTACK (also called thymus and activation-regulated chemokine or CCL27). CTACK is constitutively expressed by basal keratinocytes and is detectable on ECs and fibroblasts of the papillary dermis.11 It is thought that CTACK is mainly expressed and secreted by keratinocytes in the epidermis and captured and
Immune surveillance
Epidermis Epidermis Epidermis
CTAK (CCL27)
CCR4, CCR10
TARC (CCL17)
Superficial vascular plexus
KEY
CLA-, CCR4-, CCR10-positive lymphocyte
Arterial
5 Venular vessel releasing TARC
presented by dermal cells through glycosaminoglycans or through its receptor, CCR10 (see Fig. 9-6). CCR10 is expressed on T cells and is critical for their migration into the noninflamed skin. Knockout of CCR10 changes the balance of resident regulatory T cells (Tregs) and CD4+ effector T cells in the skin and causes overreactive inflammation after topical application of ovalbumin in a sensitized mouse. Using neutralizing anti-CTACK antibodies, the homing of CD4/ CLA/CCR10-positive T cells is blocked.11
Circulating dendritic cells may also use CLA–Eselectin interactions to extravasation into the skin. A subset of B cells also may express CLA and exhibit enhanced binding to E-selectin, potentially enabling cutaneous migration. Indeed, although very rare, B cells are present in normal human skin.12 These cells display a clonally restricted pattern, indicating recognition of a restricted antigenic repertoire—most likely against skin-associated antigens—alluding to the possibility of a skin-resident memory B-cell population. They have been shown to recirculate from the skin to regional lymph nodes by using CCR6–CCL20 interactions. The blood also contains a population of circulating T cells, characterized as effector memory T cells that function to seek out the presence of microbial antigens to which a prior immune response had been made. These, like other T cells, detect antigen using a clonally distributed repertoire of receptors that were selected to detect microbial peptides bound to class I major histocompatibility complex (MHC) molecules (human leukocyte antigen [HLA]-A, -B, or -C of CD8+ T cells) or bound to class II MHC molecules (HLA-DR, -DP, or -DQ) but not peptides derived from self-proteins. Dermal microvascular ECs express high levels of MHC molecules of both classes under basal (noninflamed conditions) and can efficiently form peptide–MHC molecule complexes. These cells also express surface proteins, such as LFA-3, 4-1BB ligand, Ox40-ligand, and ICOS-ligand, that can provide antigen-independent enhancing signals (“costimulation”) required for circulating memory T-cell activation.
A
2
Recognition of antigen on the luminal surface of ECs in an infected tissue not only activates these cells but also triggers their diapedesis across the EC lining into the skin. The cell biology of this process is unique in that T-cell recognition of antigen on the EC surface causes cells to round up, rather than spread, and cross between ECs by extending a cytoplasmic protrusion containing the microtubule organizing center and cytoplasmic granules rather than these structures following the cell body and nucleus in a trailing uropod. This antigen presenting function of ECs may be redundant with presentation of antigen by perivascular dendritic cells or macrophages that also display peptide–MHC molecule complexes on extensions that protrude into the vascular lumen. Both types of signals may supplement the initiation of immunity by resident or recirculating T cells located within the perivascular unit.
DERMAL VASCULAR ALTERATIONS OF ACUTE INFLAMMATION13
Different segments of the microvasculature contribute in different ways to acute inflammation. Historically, much of the focus has been on changes that occur in the EC lining. Local sites of inflammation are perfused at a higher level, primarily because of a shift in the balance of vasodilatory molecules released by the EC of the arterioles and, to a lesser extent, the capillaries. The principal mediator for this change is an increase in the synthesis of prostacyclin, likely resulting from a cytokine-induced increase in the expression of cyclooxygenase (COX)-2 (also known as prostaglandin H synthase 2), which is more active than constitutively expressed COX-1. The resultant increase in blood flow results in local warming (“calor”) and increased redness (“rubor”) of the inflamed site. Venular EC disrupt their intracellular adherens junctions (Fig. 9-7), allowing the extravasation of large plasma
B
123
2
proteins such as fibronectin and fibrinogen, the latter being converted to fibrin within the extravascular space, producing swelling (“tumor”). These proteins form a provisional matrix in the tissue that will allow extravasating leukocytes to attach and migrate and will serve for ultimate tissue repair. Other changes are in the display of surface molecules by the venular ECs that can serve to capture circulating leukocytes. Selectins expressed by EC cause tethering and rolling of leukocytes. Chemokines, especially IL-8 (CXCL8) for neutrophils and monocyte chemoattractant protein 1 (MCP-1, also known as CCL2), for monocytes cause spreading and firm attachment of leukocytes. Leukocyte crawling on the EC surfaces toward the extracellular junctions is mediated primarily by ICAM (intercellular adhesion molecule)-1. For mononuclear leukocytes, vascular cellular adhesion molecule (VCAM)-1 may aid in capture, rolling, and crawling. Chemokines may be made by the ECs or may be produced by sentinel cells of the perivascular unit and become displayed by attaching noncovalently to heparin sulfates on the EC luminal surface. These changes in ECs have been referred to as endothelial activation. Certain autacoids or enzymes such as thrombin may induce transient, protein synthesis– independent changes such as P-selectin translocation from intracellular storage granules (known as Weibel- Palade bodies) to the cell surface or synthesis of chemotactic lipids, such as platelet activating factor. Such changes are sometimes referred to as Type I activation, but the transient nature of such changes typically results in short-term effects, such as a wheal and flare reaction, rather than leukocyte recruitment. The latter is more dependent on new gene transcription and protein synthesis induced by inflammatory cytokines such as TNF-α, IL-1α, or IL-1β. Cytokine activation of EC can also promote coagulation by down-regulating thrombomodulin synthesis while inducing expression of tissue factor. Coagulation may be propagated by EC release of plasma membrane–derived microparticles containing tissue factor. When leukocytes reach the intracellular junctions of postcapillary venular ECs, they engage additional proteins that contribute to extravasation. Several of these, such as platelet endothelial cell adhesion molecule (PECAM)-1, poliovirus receptor, and CD99, are stored in invaginations of the lateral border plasma membrane (known as the lateral border recycling compartment) that are brought to the cell surface to engage receptors expressed on the leukocyte. (Sometimes these molecules can line short-lived transcellular channels located near but not at the actual intercellular junction, allowing leukocytes to pass through rather than between ECs. It is not known if this distinction has any physiological significance.) After the leukocyte has passed through the EC layer, it engages basement membrane proteins and PCs that express elevated levels of ICAM-1 and chemokines in response to the same cytokines that activate ECs. The interaction with PCs may extend for 30 minutes or more as PCs contract to open gaps in the basement membrane and guide extravasating leukocytes to these areas. When outside the basement membrane, extravasated leukocytes
124
Vascular changes in acute and chronic inflammation
Epidermis Epidermis Epidermis
Superficial vascular plexus
Postcapillary venules in acute inflammation are leaky; display IL-8, express E- and P-selectins, VCAM-1, ICAM-1
Postcapillary venules in chronic inflammation are less leaky; express VCAM-1, ICAM-1, E-selectin-low and release CXCL5, MCP-3, MECA-79 epitopes
KEY
Leukocyte Arteriole/Capillary Venule
continue to interact with PCs and perhaps with other cells of the perivascular unit, migrating along adventitial surface of the vessel. TNF-α and IL-1 induce proinflammatory changes in ECs that typically do not distinguish among different leukocyte types. However, the kinetics of induction of different adhesion molecules varies. Whereas E-selectin is made within the first 2 hours and correlates with the onset neutrophil recruitment, VCAM-1 is synthesized later (ie, in about 6 to 12 hours) and correlates with the onset of mononuclear leukocyte recruitment (Fig. 9-8). Polarizing cytokines, such as interferon (IFN)-γ or IL-4 and IL-13, favor the production of chemokines that preferentially activate T cells and effector cells mostly associated with TH1 or TH2-type adaptive immunity, respectively. IL-17 has little effect on ECs but instead acts on PCs to induce factors that prolong neutrophil survival (granulocyte-macrophage colonystimulating factor and granulocyte-colony stimulating factor), favoring prolongation of neutrophilic inflammation characteristic of TH17-type inflammation.14
DERMAL VASCULAR ALTERATIONS OF CHRONIC INFLAMMATION
Continuous activation of ECs is possible and results in distinct changes compared with the acute activation. Transgenic mice overexpressing TNF in an endothelium-specific manner display an inflammatory skin phenotype with epidermal hyperproliferation and
fibrosis. In culture, whereas chronic TNF-stimulated EC display a continuous and high ICAM-1 and VCAM-1 expression, E-selectin is absent and cannot be reinduced by additional TNF stimulation (ie, ECs are desensitized in regard to E-selectin expression). Interestingly, these chronic TNF-stimulated cells continuously overexpress CXCL5 and MCP-3.15 Cultured skin ECs show sustained and higher expression of E-selectin and slower internalization and degradation of E-selectin protein compared with umbilical vein endothelium, but this may be a general property of microvascular ECs.16 Nevertheless, sustained expression of E-selectin, although not unique to skin, is generally described as a skin-specific feature of chronic inflammation (see Fig. 9-8).
DERMAL VASCULAR CHANGES IN SPECIFIC DISEASES
ACUTE VERSUS CHRONIC SKIN INFLAMMATION: GENERAL CONSIDERATIONS
ACUTE VERSUS CHRONIC
SKIN INFLAMMATION:
GENERAL CONSIDERATIONS
In acute dermatitis, ECs express high levels of E- and P-selectin and moderate amounts of VCAM-1 and ICAM-1. The inflammatory infiltrate consists mainly of CLA and CD45RO-positive memory T cells. In chronic skin lesions, such as psoriasis, chronic lupus erythematosus, and mycosis fungoides, ECs express moderate amounts of E-selectin and high amounts of VCAM-1. They may also undergo morphologic changes and acquire a phenotype reminiscent of high endothelial venules. This type of EC has a cuboidal appearance, different from the flat morphology of ECs lining the other vessels. High endothelial venules express sialyted, fucosylated, and sulfated carbohydrate moieties corresponding to peripheral node addressins, which are recognized by antibody MECA-79. These addressins are constitutively expressed on high endothelial venules of lymph nodes; they bind to L-selectin expressed on leukocytes and control recirculation of T cells through lymph nodes. These sialyted, fucosylated, and sulfated carbohydrate moieties may also be expressed in chronic skin lesions such as cutaneous T-cell lymphoma.17 Also in cutaneous melanoma, blood vessels express MECA-79 moieties, and the amount of MECA-79-reactive vessels correlates with the amount of tumor infiltrating lymphocytes and with histologic signs of tumor regression.18 In squamous cell carcinoma, numbers of MECA-79–positive vessels were found to correlate with survival.
PSORIASIS
PSORIASIS
As mentioned previously, leukocytes primarily exit the blood stream through postcapillary venules. The
2
capillary loops within the papillary tips of the dermis have an arteriolar phenotype; hence, leukocytes can exit the bloodstream through the SVP below but not within the tips of the papillae. The situation is different in psoriasis. In this disease, the capillary loops elongate and change morphology. Whereas normal capillaries at the turnaround point sit on an arteriolar basement membrane, psoriatic capillary loops at the turnaround point touch the epidermis, a phenomenon called “kissing,” and remodel the basement membrane to resemble that of the postcapillary venule. ECs within the loop proliferate, producing both elongation and an increase in diameter. (The proliferation of the ECs is sometimes misinterpreted as angiogenesis, but no new vessels are actually formed.) Furthermore, the ECs of the remodeled psoriatic loop express adhesion molecules normally associated with cytokine-activated venular ECs, supporting tethering, rolling, and extravasation of leukocytes.19 Lymphocytes and neutrophils can now exit the vessel lumen within the tips of the papillae and migrate into the epidermis. This histologic picture of lymphocytes and neutrophils extravasation within the tips of the papillae has been termed “squirting papillae” and is a typical feature of psoriasis and contrasts findings in chronic eczema, in which leukocytes extravasate across post-capillary venules of the SVP (Fig. 9-9).
LICHEN RUBER PLANUS
LICHEN RUBER PLANUS
Another chronic skin disease, lichen ruber planus, has an enlarged blood microcirculatory bed within the papillary dermis compared with healthy skin. Specifically, oral lichen planus has tortuous capillaries with enlarged capillary loop diameter. Intralesional bevacizumab, anti-VEGF antibody, injections induced lesion resolution without relapse during a 3-month follow-up in a small series of patients. In this setting, vascular endothelial growth factor (VEGF) not only induces angiogenesis but is also proinflammatory by inducing endothelial adhesion molecule expression which may explain effects of the anti-VEGF antibody.
SKIN VASCULATURE IN WOUND HEALING AND CANCER
ANGIOGENESIS AND VASCULOGENESIS
ANGIOGENESIS AND
VASCULOGENESIS
In healthy adults, blood vessels are stable structures with a very slow turnover of ECs. In settings of chronic inflammation, tissue injury, or tumor growth, new vessels may be formed, and existing vessels may undergo remodeling. Much of the process of vessel formation has been learned by studying the formation of the
125
2
A
B
vascular system during embryogenesis. In embryos, ECs arise from angioblasts, which migrate to peripheral tissues and form primitive blood vessels (vasculogenesis). Subsequently, new blood vessels arise from preexisting ones (angiogenesis) (Fig. 9-10). Mesenchymal cells are then recruited into the vessel wall, where they subsequently differentiate into SMCs and PCs, a process called vascular remodeling. In adults, new blood vessels develop through angiogenesis (sprouting from preexisting blood vessels); it has been proposed that circulating endothelial progenitor cells, mesoangioblasts, and multipotent adult progenitor cells contribute to new vessel formation (adult vasculogenesis). Several investigators have proposed that these progenitors arise in the bone marrow, just as angioblasts arise
in the embryonic blood island. However, it is unclear that bone marrow–derived cells give rise to stable ECs, and many such observations have now been reinterpreted to imply a role for monocytes that express EC markers, promote angiogenesis, and then disappear as the new vessels become stable. Stem cells and endothelial progenitor cells have been identified in vessel walls as resident cells. Such cells are thought to reside in a niche, awaiting appropriate stimuli such as vessel injury to take part in the repair of damaged and the formation of new blood vessels. Among such vascular wall progenitor cells are CD34+/CD31- cells that differentiate into ECs. Other resident vascular wall progenitor cells are SMC progenitors and mesenchymal stromal cells that may arise from PCs.
Vasculogenesis and angiogenesis
Ephrin-B2 EphB4
Arterial venous Vessel segregation Pericyte recruitment Lumen formation
Angioblast Vasculogenesis
Blood islands Angiogenesis Endothelial sprouting
EGF, TGF-β FGF, VEGF ANGPT1
VEGF, ANGPT2
126
Signals inducing new vessel formation in the skin follow the principal mechanisms given in all organs. Molecules of the VEGF family—VEGF A through E— most prominently act on ECs. Fibroblast growth factor (FGF) 1 and 2 are also potent EC mitogens but also act on SMCs, PCs, and fibroblasts. In adults, FGF-1 and FGF-2 are stored in the cytoplasms of a variety of cells and are released when cells are injured, acting as “wound hormones” to stimulate local angiogenesis and connective tissue growth. Other polypeptide factors have also been found to act on ECs in vivo and in vitro, including epidermal growth factor (EGF), heparin-binding EGF–like growth factor, and hepatocyte growth factor (HGF) or scatter factor (SF). For vascular remodeling and stabilization, tyrosine kinase receptor 1 (Tie) and 2 are required. Tie-2 mediates the dialogue between mesenchymal cells and the endothelium of the immature blood vessel through binding to angiopoietins (Fig. 9-11). The eph family of receptor tyrosine kinases and their corresponding ephrin ligands are membrane bound and appear to mediate bidirectional cell-to-cell signaling. During vascular development, ephrin-B2 marks the ECs of early arterial vessels, and ephrin-B4 marks the ECs of early veins. Platelet-derived growth factor (PDGF) and transforming growth factor (TGF)-β are secreted by ECs and promote migration of
2
mesenchymal cells, matrix biosynthesis, and differentiation. Matrix glycoproteins such as fibronectin and laminin and receptors for matrix glycoproteins such as β1 and β3 integrins are also believed to play a role in vasculogenesis and angiogenesis. This seems intuitive because neither ECs nor mesenchymal cells can survive unless they interact with matrix proteins via integrin receptors.
WOUND HEALING
WOUND HEALING
Full-thickness wounds heal in interconnected and overlapping stages involving coagulation, inflammation, proliferation, and remodeling. Immediately after wounding, disrupted blood vessels contract to stop bleeding. The activation of the coagulation cascade leads to platelet clot and subsequently to fibrin clot formation. At this stage, wounded vessels function as guiding structures for circulating inflammatory cells. Complement activation and the release of CXC chemokines by platelets and keratinocytes results in the recruitment of circulating neutrophils. Surrounding capillaries become leaky, resulting in further plasma and cell accumulation within wounds. The vascular leakiness gradually decreases but persists for the
Endothelial–pericyte interactions
Lumen
Capillary
ANGPT2 acts as a partial agaonist and act to inhibit signaling by ANGPT1
ANGPT1 ANGPT2 Endothelial cell
VE-cadherin
Tie2
Signaling response No signaling response
N-cadherin
Pericyte
Shared basement membrane
127
2
next 3 to 7 days. The formation of a capillary network occurs by capillary tip extension from a parent vessel, maturation of capillary tips into capillary sprouts, anastomosis, and further branching. After this rapid formation of blood vessels during the inflammatory and proliferative stages of healing, the vascular system is remodeled, regressed, or both. The angiogenic response is mainly driven by VEGF, FGF-2, PDGF, and members of the TGF-β family (Fig. 9-12). Macrophages are the major sources of VEGF during an early healing stage; at later stages, most VEGF-expressing cells are found within the neoepithelium.20 Under conditions of injury, hypoxia, or inflammation, keratinocytes produce and release a wide range of angiogenic factors, including members of the FGF or TGF protein family, PDGF, or VEGF. Concomitantly, VEGFR-2 is upregulated on ECs, which augments endothelial responses to VEGF. FGF-2 is derived from infiltrating macrophages but also from ECs themselves. Antibodies against FGF-2 inhibit wound angiogenesis nearly completely. Stem cells from the skin stem cell niches and endogenous stem cells outside the niche support wound healing. These include hair follicle bulge and dermal sheath stem cells surrounding hair follicles, epidermal stem cells, and bone marrow–derived mesenchymal stem cells. Despite all preclinical wound-healing studies, translation of stem cell–based strategies in the clinical settings has had only limited success.21 The quantitative contribution of stem cell–derived angiogenesis compared with conventional angiogenesis through local sprouting is unknown.
TUMOR ANGIOGENESIS
TUMOR ANGIOGENESIS
Skin tumors, like any other solid tumor, acquire their blood supply from the neighboring blood capillaries. Rapid tumor growth without adequate blood supply induces hypoxia. Hypoxia is augmented because of the leaky and disorganized tumor vasculature, resulting in acute fluxes in oxygen tension and in diffusion-limited regions within the tumor. Both the tumor and the stroma compartments respond to hypoxia by expressing the hypoxia-inducible factor (HIF) family of transcription factors.22 Hypoxia stimulates tumor cells and macrophages to secrete TGF-β, PDGF, CXCL2, and endothelin, which activate fibroblasts. In fibroblasts, hypoxia stimulates extracellular matrix remodeling and the release of angiogenetic factors. Hypoxia directly effects vascular barrier function by decreasing the association between PCs and ECs. Tumors themselves may produce VEGF, FGF-2, EGF, and HGF or SF (see Fig. 9-12). Additionally, tumors may augment blood supply by intussusceptive angiogenesis (splitting of an existing blood vessel in two), vasculogenic mimicry (the formation of fluid-conducting channels by tumor cells), vessel co-option (tumor cells migrate along the preexisting vessels), and vasculogenesis.23
Vascular mimicry appears to play a specific role in metastatic melanoma (not in primary lesions) because patients with this type of vascular channel formation have a worse 5-year survival rate.24 Another interesting phenomenon observed in the tumor associated vasculature is endothelial-to-mesenchymal transition,
Vasculature in wound healing and cancer
Wound filled with fibrin containing platelets and neutrophils
VEGF, FGF-2, EGF, HGF
Skin tumor
Epidermis Epidermis Epidermis Epidermis Epidermis
VEGF, FGF-2, PDGF, TGF-β Proliferating
Macrophages
Proliferating vessels
KEY
KEY
Epidermis
VEGF, FGF-2, PDGF, TGF-β Proliferating
Macrophages
Proliferating vessels
Platelets Fibrin Arterial Venular
Macrophage Neutrophils
128
which generates cancer-associated myofibroblasts that stimulate inflammation and fibrosis and augment vascular dysfunction and as a result increase hypoxia. TGF-β is one of the driving factors of endothelial-tomesenchymal transition.25
CONCLUSIONS
The skin vascular system is unique in several respects. It is organized into functionally distinct vascular segments: the loops within the tips of the papillae, the SVP, and the DVP. These segments may individually or conjointly respond to exogenous or endogenous stimuli, thereby influencing skin disease expression. Because of their proximity to the epidermis, ECs of the SVP may directly respond to keratinocyte-derived factors such as cutaneous T cell–attracting chemokine or VEGF, which results in an altered vascular architecture, phenotype, and function. Moreover, skin EC may be directly exposed to environmental antigens. In this context, the constitutive expression of HLA class II molecules by postcapillary venules implies that ECs play a role in antigen presentation, which may increase the likelihood that the antigen is recognized in a timely manner to efficiently recruit memory T cells to sites of danger.
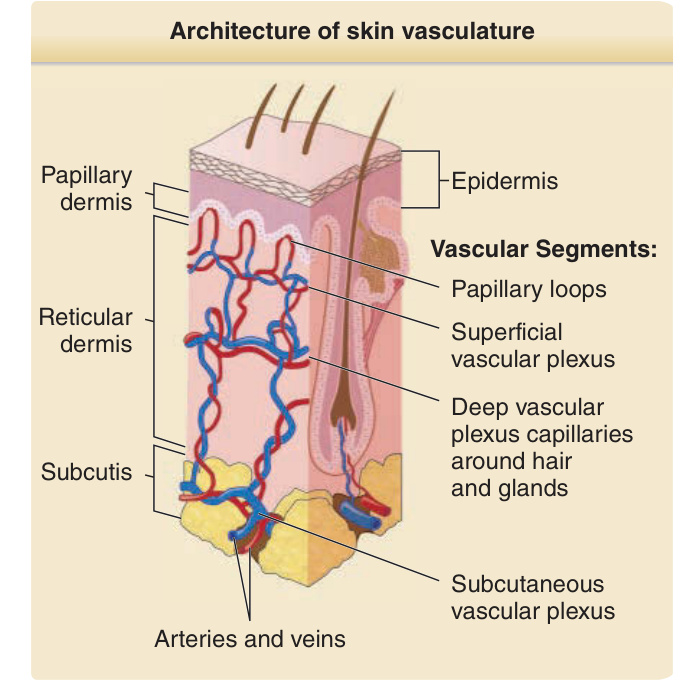
Figure 9-1 Schematic diagram of the architecture of the skin vasculature.
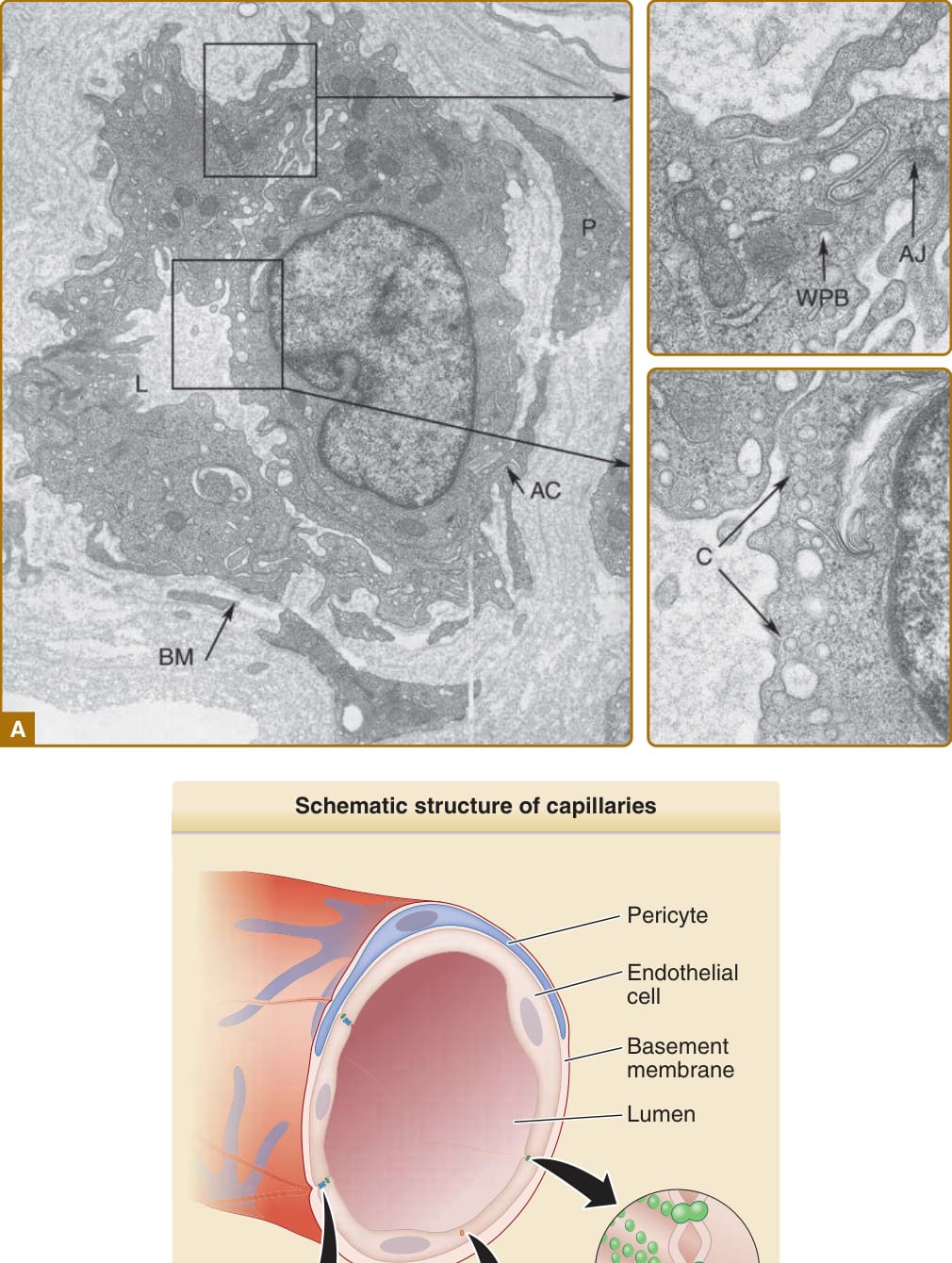
Figure 9-2 A, Ultrastructure of a postcapillary venule. AC, area of contact between endothelial cell and pericyte; AJ, adherens junction; BM, basement membrane; C, caveolae; L, lumen; P, pericyte; WPB, Weibel-Palade body. B, A schematic diagram of A.
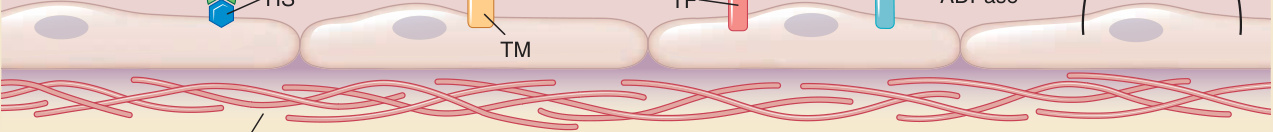
Figure 9-3 Anticoagulant and thrombotic functions of endothelial cells. To prevent coagulation, endothelial cells (1) express anticoagulant heperan sulfates (HS) that can bind and activate antithrombin III (ATIII), which then blocks the enzymatic activity of factor Xa, which converts prothrombin to thrombin, and of thrombin, which converts fibrinogen to fibrin; (2) express thrombomodulin (TM), which binds thrombin and changes it from a procoagulant enzyme that cleaves fibrinogen to fibrin into an anticoagulant enzyme that activates protein C that, in combination with protein S, then cleaves and inactivates factors V and VIII (which serve as cofactors for factors Xa and IXa, respectively); and (3) express tissue factor pathway inhibitor, which prevents tissue factor from accelerating the catalytic activity of factor VIIa, which cleaves and activates factors IX and X to factors IXa and Xa, respectively. To prevent platelet activation, endothelial cells (1) express ectoenzymes that convert extracellular adenosine triphosphate (ATP) and adenosine diphosphate (ADP), which are activators of platelets, to adenosine monophosphate (AMP), which cannot activate platelets; (2) synthesize and release inhibitors of platelet activation such as nitric oxide (NO) and prostacyclin (PGI2); and (3) prevent platelets from coming in contact with basement membrane collagen, another activator of platelet activity. These functions are mutually reinforcing as thrombin is an activator of platelets and activated platelets provide lipid surfaces that bind coagulation factors to promote the clotting cascade.
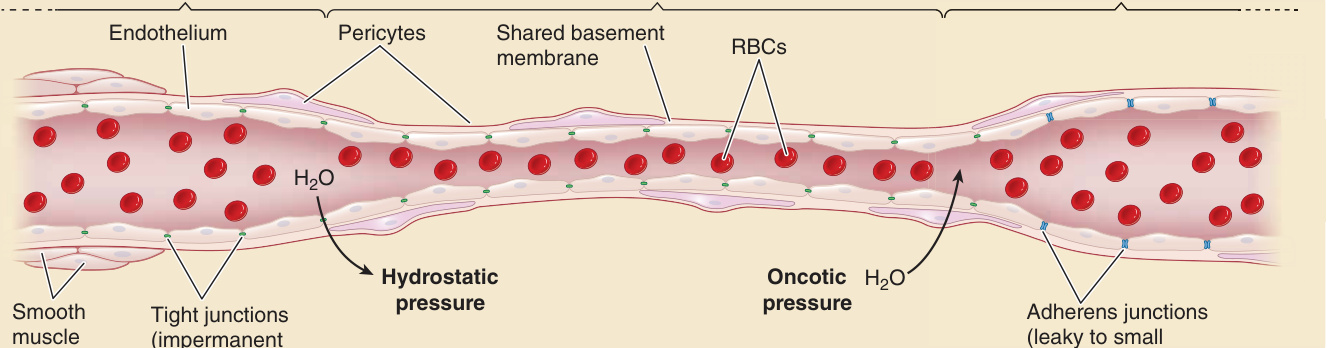
Figure 9-4 Endothelial cells establish permselective barriers. Permselectivity refers to the ability of the endothelial lining of the microvasculature to permit passage of water and solutes but not that of macromolecules. Tight junctions (TJs), which are expressed in the arterioles and capillaries, allow passage of water and solutes but prevent the passage of macromolecules and cells. Some small molecules, but not others, can pass based on size and charge. Hydrostatic pressure in the arteriolar end of the capillary forces water and solutes across the TJs and the retained macromolecules draw back water and small molecules at the venular end of the capillary through oncotic pressure. The postcapillary venules lack TJs, and their adherens junctions are dynamically regulated to allow some macromolecules to enter the tissue in the context of inflammation. RBC, red blood cell.
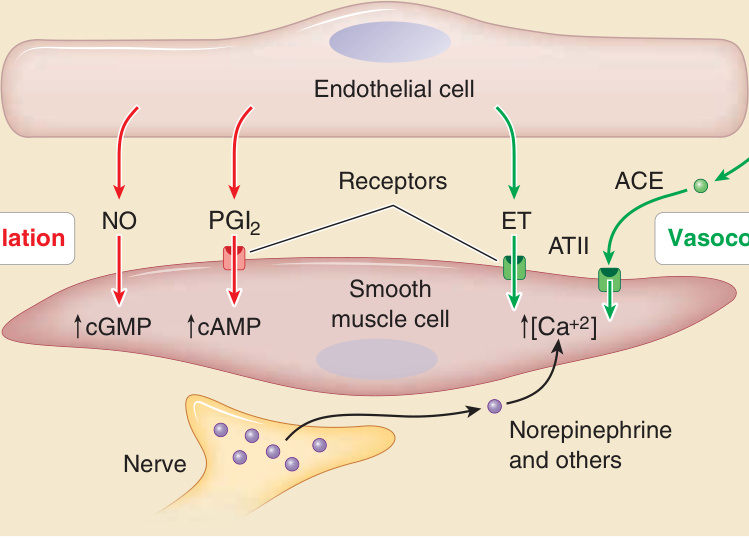
Figure 9-5 Control of perfusion by regulating smooth muscle cell tone. The active resistance of blood vessels can be modulated by altering the contractile tone mural vascular smooth muscle cells in muscular arteries and arterioles. Endothelial cells can produce vasodilators such as nitric oxide (NO) and prostacyclin (PGI2). NO directly activates soluble guanylate cyclase to elevate cyclic guanosine monophosphate (cGMP) levels, and PGI2 interacts with a membrane G–protein coupled receptor that activated adenylate cyclase to increase cycle adenosine monophosphate (cAMP). Both reduce the degree of actomyosin contraction. Endothelial cells can also produce vasocontrictors such as endothelin (ET) or, via the ectoenzyme angiotensin-converting enzyme (ACE), convert inactive angiotensin I (ATI) to vasoactive angiotensin II (ATII). Both ET and ATII signal through G protein–coupled receptors to increase the level of cytosolic free calcium ion, increasing the degree of actomyosin contraction. Nerves may release neurotransmitters that also bind to G protein–coupled receptors to increase cytosolic free calcium and actomyosin contraction. Increased actomyosin contraction shortens the length of the smooth muscle cells, which are arranged radially, thereby reducing the vessel lumen diameter and increasing resistance to flow.
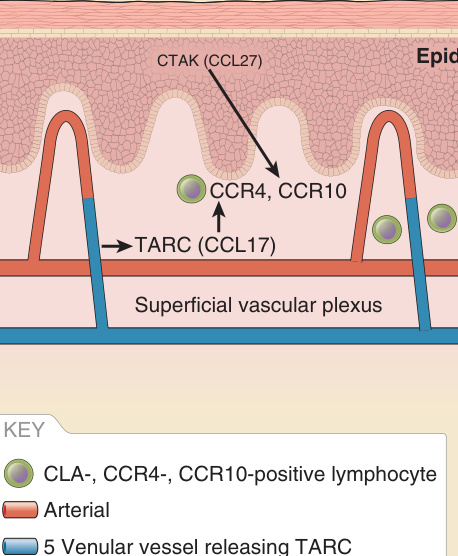
Figure 9-6 Immune surveillance.
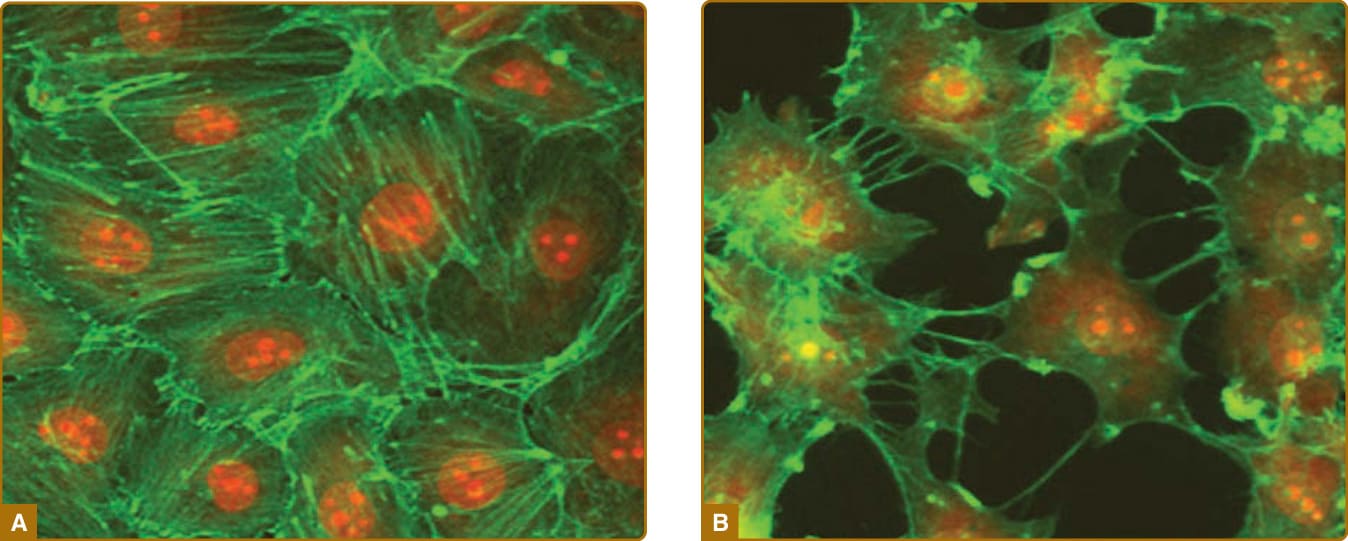
Figure 9-7 Cultured venular endothelial cells forming a continuous monolayer (A), thrombin-treated endothelial cells with disrupted intercellular junctions mimicking a leaky vessel (B). The phalloidin-staining of the actin skeleton is in green, and nuclei are in red; 1000× magnification.
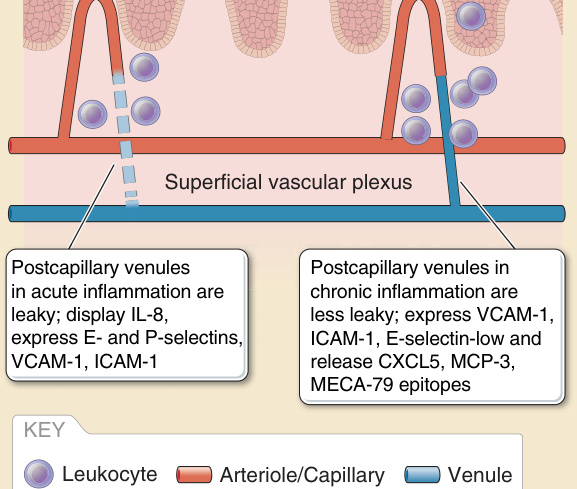
Figure 9-8 Vascular changes in acute and chronic inflammation. ICAM, intercellular adhesion molecule; IL, interleukin; MCP-3, monocyte chemoattractant protein 3; VCAM, vascular cellular adhesion molecule.
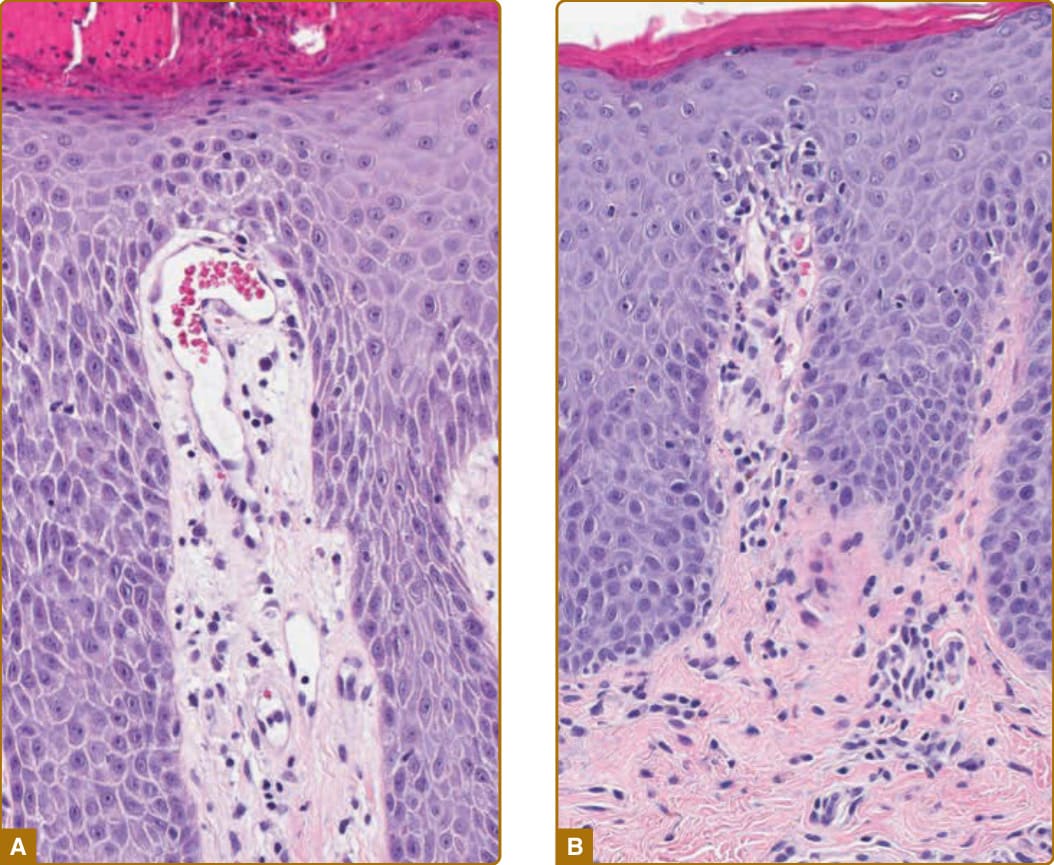
Figure 9-9 A, Vascular changes in psoriasis; the capillary loops within the tips of the papillae dilate, elongate, and at the turnaround point touch the epidermis, a phenomenon called “kissing.” B, Leukocytes exit the vessel lumen within the tips of the papillae (“squirting papillae”); 20× magnification.
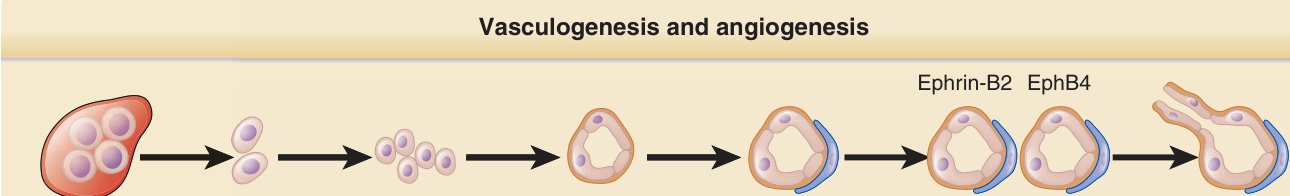
Figure 9-10 Vasculogenesis and angiogenesis. ANGPT, angiopoietin; FGF, fibroblast growth factor; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor.
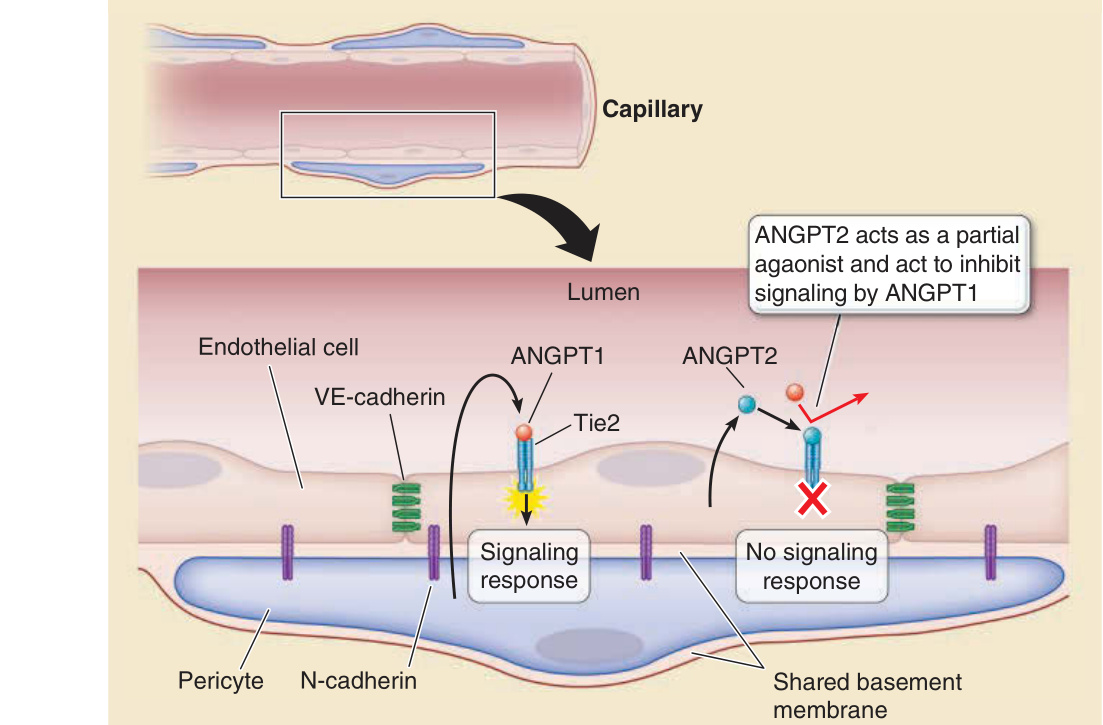
Figure 9-11 Endothelial cell (EC)–pericyte interactions. Pericytes are localized to within the basement membrane of the EC lining of microvessels and contribute to its synthesis and maintenance, and the composition and organization of the basement membrane may modify EC functions. Pericytes directly contact ECs, forming adherens junctions involving N-cadherin linkages. Pericytes also stabilize ECs by paracrine signals; the best understood of which involves pericyte secretion of angiopoietin 1 (ANGPT1), which binds to and activates the receptor tyrosine kinase Tie2 expressed on the endothelium. ANGPT1 binding prevents EC-derived ANGPT2 from binding to the same receptor. ANGPT2 acts as a partial agonist, and when its levels increase, it prevents ANGPT1-mediated signaling, leading to disruption of endothelial cell junctions associated with capillary leak, angiogenesis, or both.
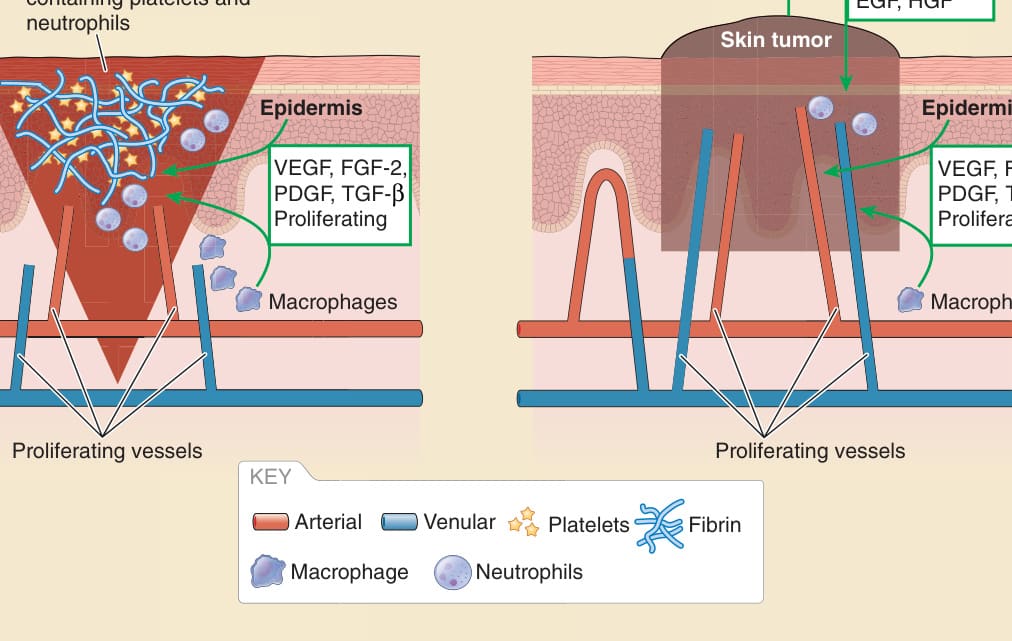
Figure 9-12 Vasculature in wound healing and cancer. EGF, endothelial growth factor; FGF, fibroblasst growth factor; HGF, hepatocyte growth factor; PDGF, platelet-derived growth factor; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor.

TABLE 9-1 Properties and Functions of Normal Skin Vasculature

TABLE 9-2 Thermoregulation in Nonglabrous and Glabrous Skin