自體發炎性疾病 (Autoinflammatory Disorders)
總論
- 自體發炎性疾病 (autoinflammatory disorders) 為先天性免疫系統 (innate immune system) 失調所致;發炎為無菌性 (sterile),但症狀(發燒、皮膚病灶、關節痛)類似感染。
- 不具自體免疫 (autoimmunity) 的標誌(無高自體抗體效價、無抗原特異性 T 細胞)。
- 介白素 (interleukin, IL)-1 在大部分單基因 (monogenic) 疾病中扮演要角;多於兒童期(有時新生兒期)發病,以體染色體隱性 (autosomal-recessive) 或顯性 (autosomal-dominant) 遺傳,亦有偶發性 (sporadic) 病例。
- 特徵性皮膚疹:嗜中性球性蕁麻疹樣病灶、膿疱症、蜂窩性組織炎樣病灶、紅斑性與斑丘疹性疹。
- 已有特定治療:IL-1 拮抗劑(CAPS、Schnitzler、DIRA);秋水仙鹼 (colchicine)(FMF)。
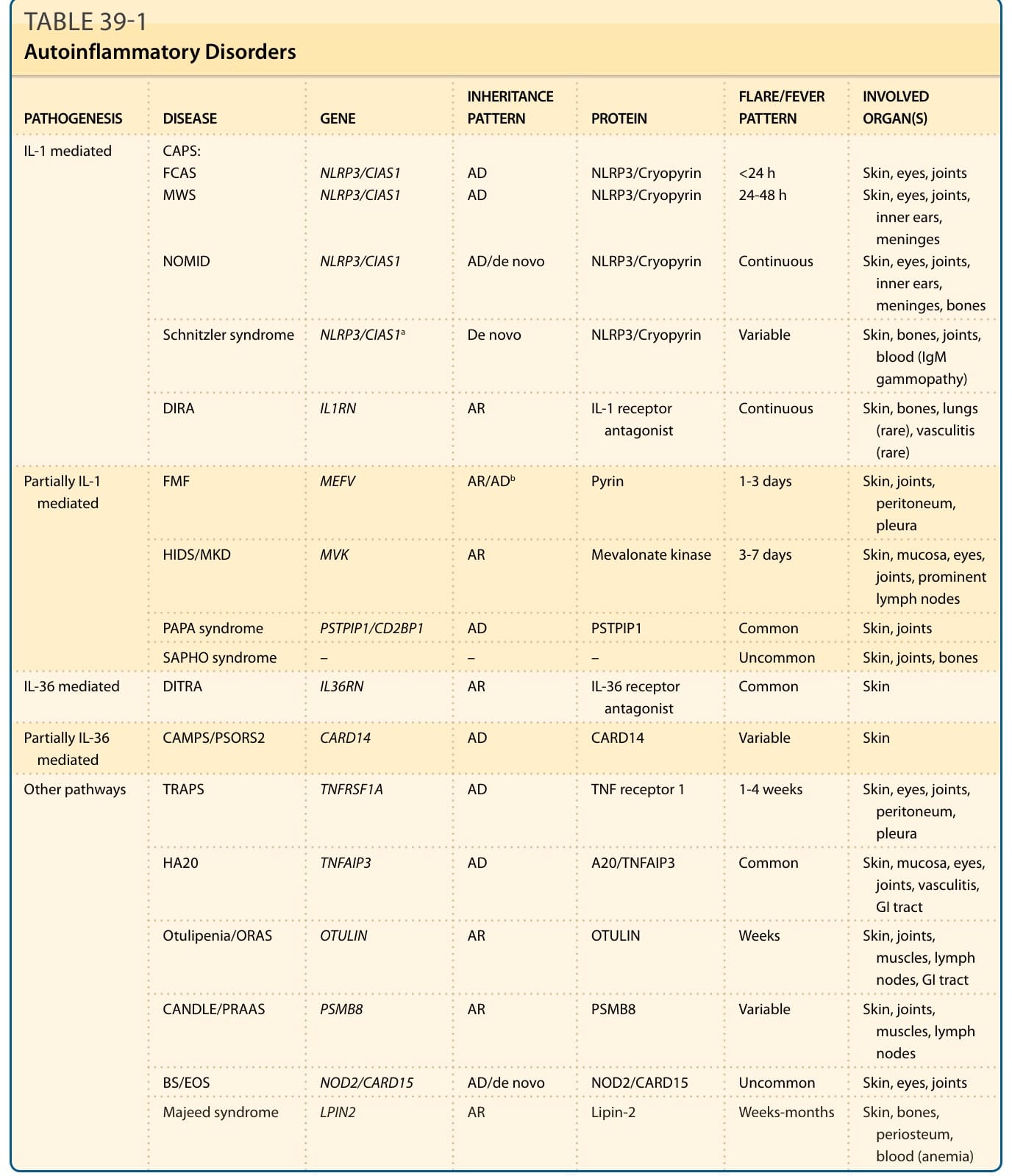
表 39-1:自體發炎性疾病(致病機轉、基因、遺傳模式、發燒模式與侵犯器官總表)
冷凝素相關週期性症候群 (CAPS)
- 罕見體染色體顯性疾病,由 NLRP3(編碼冷凝素 cryopyrin)功能獲得 (gain-of-function) 突變→發炎體 (inflammasome) 活化→異常 IL-1β 分泌。盛行率約每百萬人 1–2 人。
- 疾病譜(嚴重度由輕至重):家族性冷自體發炎性症候群 (FCAS)、Muckle-Wells 症候群 (MWS)、慢性嬰兒型神經皮膚關節症候群 (CINCA)/新生兒發病多系統發炎性疾病 (NOMID)。FCAS、MWS 為家族性;CINCA/NOMID 為偶發性。
- 臨床表現:蕁麻疹樣皮膚病灶為共同且最初徵象,通常不癢、對抗組織胺 (antihistamines) 無效。
- FCAS:低溫暴露後 1–2 小時發作,<24 小時,關節痛、結膜炎;冰塊激發試驗陰性。
- MWS:三聯徵為蕁麻疹、耳聾、類澱粉沉積症 (amyloidosis);漸進性感覺神經性聽力喪失(第二至三個十年)、無菌性腦膜炎、葡萄膜炎。
- CINCA/NOMID:出生即瀰漫性紅斑與發燒、慢性無菌性腦膜炎、腦萎縮、視乳突水腫、長骨骨骺過度生長 (epiphyseal overgrowth)、身材矮小。
- 組織學:淺層與真皮中層血管周圍嗜中性球浸潤,無血管炎,表皮完整。
- 診斷:臨床史為主+急性期反應物 (CRP、ESR、SAA) 升高、白血球增多;NLRP3 基因檢測確認,惟陰性不能排除(可能為鑲嵌性 mosaicism)。長骨 X 光骨骺過度生長為 CINCA/NOMID 獨有。
- 鑑別:後天性冷蕁麻疹、新生兒敗血症/TORCH、全身性幼年型特發性關節炎 (sJIA, Still 病)。
- 處置:IL-1 拮抗劑為標準照護、反應戲劇化。
- Anakinra(Kineret,重組 IL-1 受體拮抗劑)每日皮下注射;FCAS 約 0.5 至 1.5 mg/kg/day,CINCA/NOMID 可能需 6 至 10 mg/kg/day,可通過血腦障壁。
- Rilonacept(Arcalyst,IL-1 Trap)半衰期 8.6 天,每週皮下 160 mg(成人)。
- Canakinumab(Ilaris,抗 IL-1β 單株抗體)平均半衰期 26 天,每 8 週皮下 150 mg(成人)。需終生治療、盡早開始以減少不可逆神經併發症。
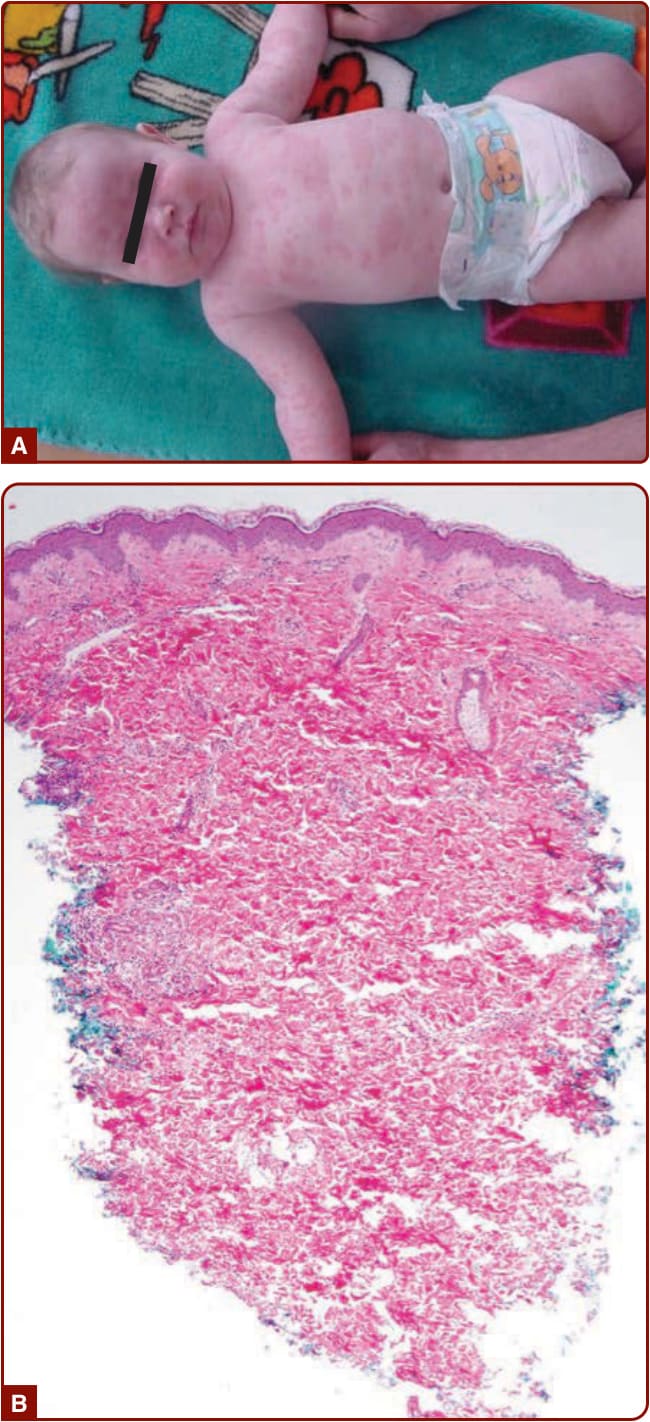
圖 39-1:CAPS。A,MWS 嬰兒的蕁麻疹樣皮膚病灶;B,組織學顯示血管周圍嗜中性球浸潤而無血管炎。
Schnitzler 症候群
- 罕見晚發型疾病(約 50 歲後),視為偶發性後天性自體發炎性疾病;全球約 300 例。部分病人有限於骨髓系的 NLRP3 突變鑲嵌性。
- 臨床:反覆蕁麻疹樣病灶(最初徵象,可早數年;不癢、24–48 小時消退、抗組織胺無效)、間歇性發燒(>40°C)、骨痛、關節痛、淋巴結病變、肝脾腫大。組織學同 CAPS(嗜中性球浸潤、無血管炎)。
- 特徵:85% 有單株 IgM 球蛋白病變(多為 IgMκ),為與 CAPS 區別之關鍵。
- 鑑別:蕁麻疹性血管炎(有纖維素樣壞死,Schnitzler 無)、冷凝球蛋白血症、成人型 Still 病、HIDS。
- 病程:約一至二成發展為淋巴增生性疾病(Waldenström 巨球蛋白血症、B 細胞淋巴瘤)。
- 處置:抗組織胺無效;anakinra 與 canakinumab 可在逾九成病例誘導完全緩解,停 anakinra 後 1 至數天復發。
IL-1 受體拮抗劑缺乏症 (DIRA)
- 極罕見體染色體隱性,IL1RN 突變(始祖突變)→IL-1Ra 缺失/功能障礙→無拮抗的過度活躍 IL-1 訊號。
- 臨床:圍產期至數月齡發病的膿疱性皮膚疹、口腔黏膜病灶、體重增加不良、疼痛性關節腫脹;高燒非典型。皮膚表現變異大(正常皮膚帶膿疱至全身性嚴重膿疱症或魚鱗癬樣病灶)。放射學:前肋骨末端氣球狀增寬、長骨骨膜抬升、多灶性溶骨性病灶。組織學:表皮內嗜中性球與膿疱、乳突真皮水腫。
- 診斷:易誤為感染性骨髓炎(培養陰性、抗生素無效);IL1RN 基因檢測確認。
- 病程:未治療→嚴重發炎反應症候群與多器官衰竭致新生兒死亡。
- 處置:重組 IL-1Ra(anakinra/Kineret)替代缺失蛋白,反應快速戲劇化,需終生治療。
IL-36 受體拮抗劑缺乏症 (DITRA)
- 罕見體染色體隱性,IL36RN 同型合子或複合異型合子突變→IL-36 訊號異常→角質細胞 (keratinocytes) 過度產生 IL-8(嗜中性球趨化劑)。為單基因型全身性膿疱性乾癬 (GPP)。
- 臨床:反覆突發的全身性紅斑膿疱性皮膚疹+高燒(40–42°C)、嗜中性球增多。誘發:病毒/細菌感染、藥物(如 amoxicillin)、月經、懷孕、停用類視色素 (retinoid)。無關節侵犯,可有指甲失養。組織學:上層表皮 Kogoj 海綿狀膿疱合併棘層肥厚。
- 鑑別:細菌性膿痂疹、角質層下膿疱性皮膚病 (Sneddon-Wilkinson)、疱疹樣膿痂疹、急性全身性發疹性膿疱症(部分亦帶 IL36RN 突變)。
- 處置:最佳治療未定;IL-1 與 IL-17 拮抗劑有療效。
CARD14 介導的膿疱性乾癬/乾癬 2 (CAMPS/PSORS2)
- 罕見體染色體顯性,CARD14(CARMA2)功能獲得突變→MAPK 與 NF-κB 訊號異常→過度產生 IL-8 與 IL-36γ。
- 臨床:各種乾癬症狀,含斑塊型與膿疱性乾癬。
- 處置:未定;曾有 1 例以 IL-12/IL-23 拮抗劑 ustekinumab 成功治療。
家族性地中海熱 (FMF)
- 最常見且第一個經基因鑑定的單基因自體發炎性疾病;多為體染色體隱性(亦有少見顯性型),MEFV(編碼 pyrin)突變→pyrin 發炎體活化→異常 IL-1β 分泌。
- 好發地中海族群(亞美尼亞、西班牙裔猶太、阿拉伯、土耳其),此族群中可能每 200 人 1 人發病;80% 於 10 歲前、90% 於 20 歲前診斷。
- 臨床:反覆短暫發熱性發作(持續 1 至 3 天,隨機發生,發作間完全無恙)+腹膜炎、胸膜炎、滑膜炎、丹毒(蜂窩性組織炎)樣皮膚病灶。後者為界線分明、溫熱、觸痛、腫脹的紅色病灶,主要於小腿前側、足背或踝部。誘發:低溫、疲倦、情緒壓力、月經。組織學:淺層真皮水腫+血管周圍與間質嗜中性球浸潤,無血管炎。
- 診斷:臨床為主;發作時 ESR、CRP、SAA、纖維蛋白原升高、低度嗜中性球增多;MEFV 基因檢測可確認(非全部可測得)。
- 鑑別:急性闌尾炎、sJIA(Still 病,每日發燒不消退)、感染性丹毒/蜂窩性組織炎、TRAPS、HIDS、CAPS。
- 病程/預後:終生疾病、不可治癒;最嚴重併發症為繼發性類澱粉沉積症→腎衰竭(主要死因),秋水仙鹼可顯著降低其發生率。
- 處置:秋水仙鹼為所有病人建議用藥(預防發作、最小化亞臨床發炎、預防類澱粉沉積症進展);不耐受/無效或腎功能受損者第二線為 IL-1 拮抗劑(含 canakinumab)。
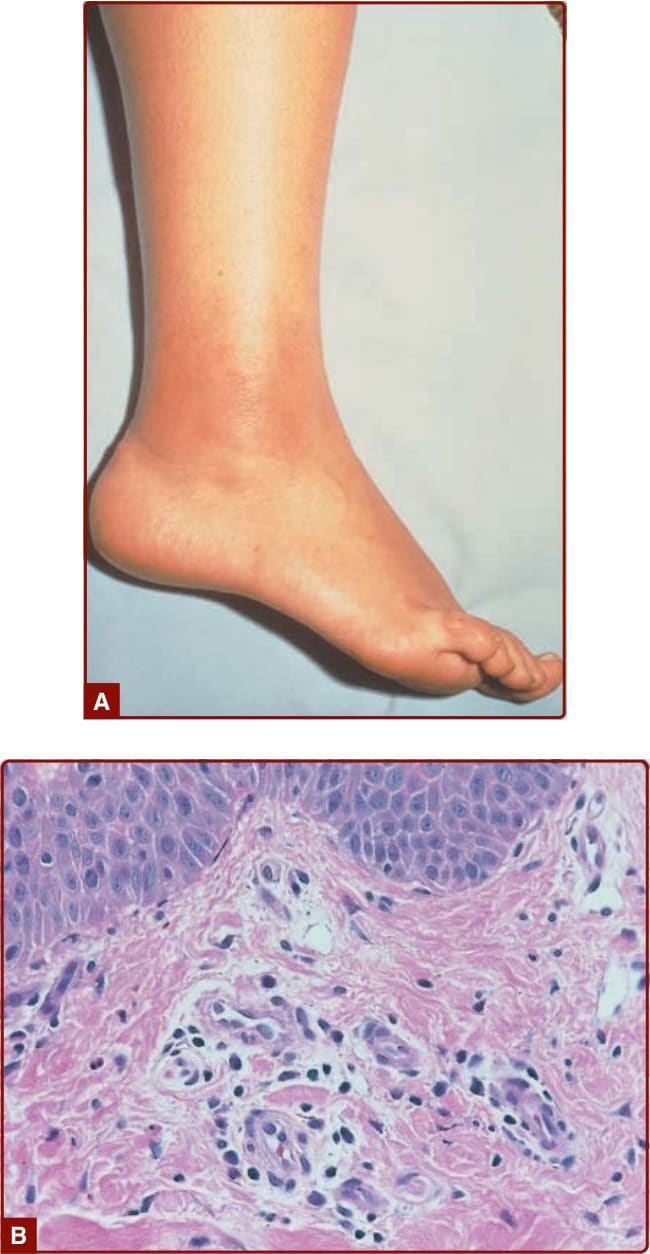
圖 39-6:FMF。A,右踝的丹毒樣皮膚病灶(界線分明、腫脹、鮮紅);B,組織學顯示淺層真皮血管周圍發炎性浸潤。
高免疫球蛋白 D 合併週期性發燒症候群/甲羥戊酸激酶缺乏症 (HIDS/MKD)
- 罕見體染色體隱性,MVK(編碼甲羥戊酸激酶 mevalonate kinase,參與膽固醇與類異戊二烯生合成)突變→酵素活性降至正常 1%–10%→經複雜機轉過度產生 IL-1β。好發荷蘭(每 350 人 1 人帶隱性突變)與法國北部,多於第一年發病。
- 甲羥戊酸尿症 (mevalonic aciduria) 為酵素活性 <1% 的嚴重致命型(週期性發燒、嚴重神經損傷與生長遲滯、早期死亡)。
- 臨床:高、尖峰式發燒(前有畏寒)+腹痛、腹瀉、嘔吐、頭痛、關節痛、頸部淋巴結病變、皮膚病灶;半數有口腔口瘡性潰瘍(有/無生殖器潰瘍)。發作持續 3 至 7 天,自發停止,每 4 至 8 週復發。誘發:免疫接種、創傷(含手術)、壓力。最常見皮膚病灶為廣泛紅斑性斑疹與丘疹(肢端>軀幹)。組織學:非特異性血管周圍淋巴球浸潤帶部分嗜中性球。
- 診斷:臨床懷疑+基因檢測確認;最典型為血清 IgD 持續升高(約 20% 可正常),80% 亦 IgA 升高(IgD 升高非特異)。
- 鑑別:闌尾炎、FMF(秋水仙鹼有效可助鑑別)、TRAPS、MWS、貝賽特氏病。
- 病程:長期預後良好,併發症罕見(類澱粉沉積症為例外且罕見)。
- 處置:無單一特定治療;NSAIDs 緩解發燒;難治者發作初口服糖皮質素 (oral glucocorticoids);秋水仙鹼無效。Anakinra、TNF-α 抑制劑 etanercept 部分成功;canakinumab(每 4 至 8 週皮下)於 2016 年成為首個 FDA 核准用於 HIDS 的生物製劑。
腫瘤壞死因子受體相關週期性症候群 (TRAPS)
- 罕見體染色體顯性,TNFRSF1A(編碼 TNFR1)突變;致病機轉未充分理解(TNFR 脫落受損、配體結合減少、細胞凋亡訊號缺損、TNFR1 異常寡聚化)。繼 FMF 後第二常見遺傳性週期性發燒症候群;原稱家族性愛爾蘭熱。約半數無家族史,多於 10 歲前發病。
- 臨床:反覆高度發燒(伴畏寒)通常持續 3 週(可數天至數月、每 6 週至每數年復發)。誘發:感染、輕微損傷、壓力、運動。伴無菌性腹膜炎之腹痛、由單核球性筋膜炎造成的移行性肌痛、漿膜炎、單關節炎、結膜炎、眼周水腫。皮膚:最常為紅斑性斑塊(可移行、與下方肌痛相關)。組織學非特異性(血管周圍淋巴球與單核球浸潤)。
- 診斷:臨床+發作時 ESR、CRP、SAA、纖維蛋白原升高與白血球增多;可溶性 TNFR 超家族 1A 血清濃度可偏低;基因檢測確認。
- 病程:約 10%–15% 因 SAA 沉積發生類澱粉沉積症→蛋白尿→腎衰竭。
- 處置:NSAIDs;高劑量皮質類固醇通常有效但副作用;etanercept 部分有效但可能誘發嚴重發炎;anakinra 與 canakinumab 更有效,canakinumab 於 2016 年獲 FDA 核准。
A20 單倍體不足 (HA20)
- 罕見體染色體顯性,TNFAIP3(A20,泛素編輯酵素)功能喪失突變→單倍體不足→K63 泛素化增加與 NF-κB 依賴性促發炎細胞激素增加。表現與貝賽特氏病 (BD) 無法區分,視為 BD 疾病譜一部分。
- 臨床:兒童或早成年期發病;發燒、口腔與生殖器雙極性潰瘍、皮膚病灶(紅斑性丘疹、假性毛囊炎、結節性紅斑樣病灶、針刺反應 pathergy)、葡萄膜炎、多關節炎;可有視網膜血管炎、胃腸道潰瘍、CNS 血管炎及多種自體抗體。
- 處置:秋水仙鹼部分有效;TNF-α 與 IL-1R 拮抗劑療效不一。
Otulipenia/OTULIN 相關自體發炎性症候群 (ORAS)
- 罕見體染色體隱性,OTULIN(去泛素酶,切割線性泛素鏈負向調節 NF-κB)功能喪失突變→線性泛素化增加與促發炎轉錄本升高。已報告 6 名病人(皆近親婚配後代)。
- 臨床:出生數天至數月起的嚴重全身性發炎—長時間發燒、皮疹、關節痛、腹瀉、漸進性脂肪失養 (lipodystrophy)、發展遲緩;常見伴嗜中性球浸潤的復發性結節性脂膜炎。組織學:嗜中性球性皮膚炎與脂膜炎。
- 處置:TNF-α 抑制劑可治療(3 名反應良好);全身性類固醇與 anakinra 療效不一或無效;6 名中 2 名(皆未用 TNF-α 抑制劑)死亡。
CANDLE/PRAAS
- 罕見體染色體隱性,PSMB8(蛋白酶體次單元 β8)同型合子或複合異型合子突變(亦有雙基因遺傳)→蛋白酶體活性失調與第 I 型干擾素 (interferon) 訊號增加。涵蓋中條-西村症候群等舊名,約 60 例。
- 臨床:第一年發病;反覆發燒、凍瘡樣 (pernio-like) 與結節性紅斑樣疹、細長杵狀手指合併關節攣縮、漸進性脂肪肌肉萎縮(主要臉部與上肢);可有眼周紅斑水腫、肌炎、基底神經節鈣化。
- 診斷:慢性貧血、CRP/ESR 升高、不固定高球蛋白血症與自體抗體;組織學為真皮與皮下脂肪緻密單核細胞、組織球、嗜酸性球與嗜中性球浸潤,無白血球碎裂性血管炎;PSMB8 基因檢測確認。
- 處置:迄今無有效治療;全身性類固醇僅暫時改善皮疹、減量後復發;多種抗風濕/免疫抑制藥(anakinra、tocilizumab、TNF-α 抑制劑等)無效或僅短暫改善。
Blau 症候群/早發型類肉瘤病 (BS/EOS)
- 罕見;BS 為體染色體顯性,EOS 為其偶發性形式;NOD2(CARD15,辨識胞壁醯二肽)功能獲得突變,致病機轉大多未知。
- 臨床:三聯徵為肉芽腫性皮膚炎、關節炎、葡萄膜炎,典型 4 歲前發病。皮膚為無症狀、略脫屑、黃至棕紅平頂丘疹(>90% 病人);無類肉瘤病常見的雙側肺門淋巴結病變。約 95% 有關節炎—慢性、對稱、大多無痛之多關節炎(關節內滑膜炎與腱鞘炎)。葡萄膜炎可進展為全葡萄膜炎,20%–30% 顯著視力喪失。
- 組織學:上層真皮由組織球與多核巨細胞組成的非乾酪性肉芽腫 (noncaseating granulomas)。
- 診斷:切片證實非乾酪性肉芽腫+NOD2 基因檢測;家族史為 BS 關鍵;肌肉骨骼超音波有用。
- 處置:證據不足;靜止期低劑量皮質類固醇通常滿意;葡萄膜炎與關節炎常合併全身性類固醇、免疫抑制劑與生物製劑(多為 TNF 拮抗劑);methotrexate 可抑制關節炎並助類固醇減量。
化膿性關節炎、壞疽性膿皮症與痤瘡症候群 (PAPA)
- 罕見體染色體顯性,PSTPIP1(CD2BP1)突變;可與 pyrin 結合增加→經發炎體誘導 IL-1β 過度產生(確切機轉未明,秋水仙鹼無效)。
- 臨床:無菌性化膿性關節炎(第一個十年起、進行性破壞、發作後富嗜中性球之化膿性滑膜發炎)+青春期出現的壞疽性膿皮症(癒合不良潰瘍、潛行性邊緣、常於損傷部位)與嚴重結節囊腫性痤瘡。發燒罕見。
- 診斷:臨床懷疑+發作時急性期反應物升高;PSTPIP1 基因檢測確認。
- 鑑別:sJIA、FMF、SAPHO、Majeed 症候群。
- 處置:具挑戰性,反應差異大;嚴重化膿性關節炎可引流+關節內類固醇;高劑量皮質類固醇對關節炎與壞疽性膿皮症佳但惡化痤瘡;TNF-α 拮抗劑 (etanercept) 與 IL-1Ra (anakinra) 部分有效。
Majeed 症候群
- 罕見體染色體隱性,LPIN2(編碼 lipin-2)突變,機轉未充分理解;僅少數中東家族。
- 臨床三聯徵:慢性復發性多灶性骨髓炎 (CRMO)、先天性紅血球生成異常性貧血 (CDA)、類似 Sweet 症候群的嗜中性球性皮膚病。CRMO 嬰兒/兒童早期起反覆疼痛與關節腫脹;CDA 第一年起低色素小球性貧血(輕度至輸血依賴)。組織學似 Sweet 症候群(真皮嗜中性球浸潤、乳突真皮水腫、無血管炎)。
- 診斷:臨床+LPIN2 基因檢測;X 光溶骨性病灶伴硬化;切片排除感染(培養無菌)。
- 處置:CRMO 用 NSAIDs,無效則皮質類固醇;對 etanercept 無效者改用 anakinra 或 canakinumab 反應戲劇化;CDA 視需要紅血球輸血;皮膚病用短療程口服皮質類固醇。
滑膜炎、痤瘡、膿疱症、骨質增生與骨炎症候群 (SAPHO)
- 發炎性疾病,特徵為反覆嗜中性球性皮膚與骨關節 (osteoarticular) 表現;多因子病因(遺傳、環境、免疫),多為偶發性。常見於中年成人。
- 臨床:皮膚與骨關節表現多於彼此 2 年內出現。皮膚以掌蹠膿疱症 (PPP) 最常見,其次嚴重痤瘡(聚合性、暴發性痤瘡、化膿性汗腺炎)。骨關節以寡關節炎(胸肋、胸鎖、薦髂、膝、踝)與前胸壁骨炎為主。骨骼切片顯示無菌性骨髓炎,後期被硬化性骨小樑與骨髓纖維化取代。
- 鑑別:感染性骨髓炎、骨腫瘤(尤文氏肉瘤、骨母細胞瘤)、乾癬性關節炎;兒童另含蘭格罕細胞組織球增生症、DIRA、PAPA、Majeed。
- 處置:以症狀治療為主;NSAIDs 第一線;關節內/全身性皮質類固醇暫時有效;PPP 與膿疱性乾癬對局部類固醇與 PUVA 反應佳;雙磷酸鹽對骨骼病灶有時很有效;難治用 TNF-α 拮抗劑(可能矛盾性惡化);anakinra 於病例報告顯示前景。