惡性萎縮性丘疹病 (Malignant Atrophic Papulosis, Degos Disease) 精華筆記
疾病定義與分類
- 惡性萎縮性丘疹病(malignant atrophic papulosis,又稱 Degos 病 Degos disease)為罕見的原發性血管阻塞性疾病 (primary vasoocclusive disorder),主要侵犯皮膚、胃腸道 (GI tract) 與中樞神經系統 (CNS)。
- 其特徵性病灶實為皮膚血栓閉塞性血管病變 (cutaneous thromboobliterative vasculopathy) 的標記,而非單一特定疾病。可見於至少 2 種情境:
- (a) 特發性疾病:典型 Degos 病或其良性變異型 (benign variant)。
- (b) 結締組織疾病的替代性臨床表現:如抗磷脂質症候群 (antiphospholipid syndrome)、紅斑性狼瘡 (lupus erythematosus)、皮肌炎 (dermatomyositis)、系統性硬化症 (systemic sclerosis)。
流行病學
- 罕見,文獻約 250 例;幾乎都發生於白人,第三至第四個十年(30–40 歲)最常見,可發生於任何年齡。
- 男性多於女性(3:1)。多為散發性,家族性病例多符合體染色體顯性 (autosomal dominant) 遺傳。
致病機轉
- 病因不明,組織病理提示原發性血管阻塞,以血管凝血病變 (vascular coagulopathy) 及/或內皮細胞損傷為主要機轉。
- 廣泛研究促血栓因子 (prothrombotic factors) 未能反覆找到單一異常;部分病人有纖維蛋白溶解 (fibrinolysis) 受抑制與血小板異常。
- Magro 等人提出 Degos 病可能為干擾素-α (interferon-α) 介導的內皮病變,補體攻擊複合體 C5b-9 (complement attack complex C5b-9) 可能促成血栓性微血管病變;但補體損傷可能為晚期事件,故抗 C5 抗體 eculizumab 能搶救急性重症卻對閉塞性纖維內膜動脈病變無效。
臨床表現
- 起初為成簇 (crops) 出現、2 至 10 mm、大致無症狀的玫瑰色斑 (rose-colored macules),快速進展為圓頂狀堅實丘疹,部分有中央臍狀凹陷或壞死。
- 數天至數週內演變為瓷白色 (porcelain-white) 萎縮性丘疹,邊緣帶玫瑰色紅斑及/或微血管擴張 (telangiectasia),極似白色萎縮 (atrophie blanche);終留下水痘樣 (varicelliform) 白色疤痕。
- 好發於軀幹與四肢;手掌、足底、顏面、頭皮、生殖器通常不受侵犯。僅局限於肢端 (acral) 較提示結締組織疾病而非 Degos 病。
- 眼部可受侵犯,最常見為結膜上的無血管斑 (avascular patch)。
- 典型 Degos 病:皮膚病灶數個至超過 100 個,通常先於系統性表現;GI 表現含腸穿孔與腹膜炎,CNS 表現含出血性或缺血性中風;屍檢可見多器官小血管血栓侵犯。
- 良性 Degos 病:僅皮膚侵犯,多數家族性病例屬良性;典型型與良性型無明確區分,無法預測誰會發展皮膚外侵犯。
- 次發性 Degos 病樣病灶:見於紅斑性狼瘡、皮肌炎、系統性硬化症、多血管炎性肉芽腫病 (Wegener granulomatosis)、克隆氏病 (Crohn disease)、類鴉片誘發血栓性微血管病變、小病毒 B19 (parvovirus B19) 病毒血症等。
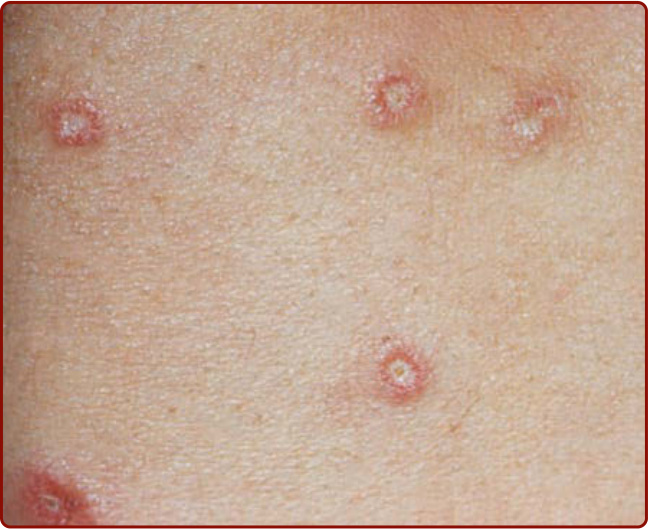
圖 146-4:惡性萎縮性丘疹病典型瓷白色中央與紅斑性隆起邊緣的丘疹。
診斷與鑑別診斷
- 組織病理:細胞稀少 (cell-poor)、楔形 (wedge-shaped) 真皮壞死區,伴真皮水腫與大量黏液 (mucin) 沉積;病灶基部可見閉塞血管伴偶發血栓、內膜纖維化 (intimal fibrosis)、增生內皮細胞與稀疏血管周圍淋巴球浸潤。從未見完全發展的白血球破碎性嗜中性球性血管炎,故不應歸類為血管炎。
- 因部分發現(黏液浸潤、介面性皮膚炎、血管周圍淋巴球浸潤)與紅斑性狼瘡、皮肌炎共有,有人認為 Degos 病可能是紅斑性狼瘡的變異型。
- 無診斷性或預後性標記;每位病人應查抗核抗體、狼瘡抗凝物 (lupus anticoagulant)、抗心磷脂質抗體 (anticardiolipin antibodies),並篩檢抗磷脂質抗體與冷凝球蛋白 (cryoglobulins)。突然發作者應以 PCR 尋找小病毒 B19 病毒 DNA。
- 診斷通常臨床確立,但應始終執行確認性切片 (confirmatory biopsy)。
- 鑑別診斷:常見為白色萎縮 (atrophie blanche);務必排除伴 Degos 病樣病灶的結締組織疾病、克隆氏病、多血管炎性肉芽腫病、類鴉片誘發血栓性微血管病變、小病毒 B19 感染。
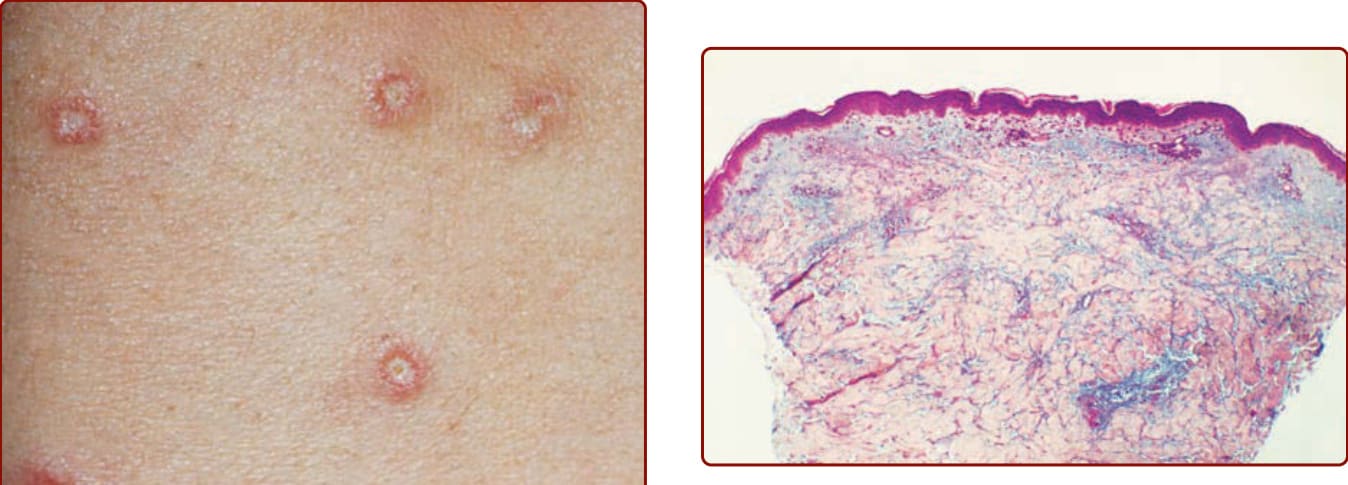
圖 146-5:楔形、細胞稀少的真皮壞死區伴大量黏液沉積(hematoxylin-eosin-saffron-Astra blue 染色,×40)。
併發症
- GI 出血、穿孔與腹膜炎為最常見併發症及主要凶險事件,在罹患系統性型疾病的百分之三十病人中有高達百分之七十三會發生。
- 神經學併發症較少見:半身輕癱、失語症、多條腦神經侵犯、單肢癱瘓、感覺障礙、癲癇發作;罕見可因進行性神經侵犯致死(尤其兒童)。
- 系統性疾病病人 5 年存活率約百分之五十四。
預後與臨床病程
- 約百分之三十病人於診斷後 2 至 3 年內出現皮膚外表現;但系統性侵犯可在皮膚病灶後多年才發生,結果難以預測。
- 若診斷後 7 年仍無皮膚外表現,呈良性單症狀皮膚病程的機率大於百分之九十。
- 就診時即有皮膚外侵犯者致死率大於百分之五十,多在 2 至 3 年內死亡(主因嚴重腸道侵犯)。家族性 Degos 病預後較佳。
治療
- 無對照臨床試驗,僅零星個案報告;因部分面向類似類網狀青斑性血管病變 (livedoid vasculopathy),可借鏡其療法。
- 應戒菸、篩檢並控制心血管危險因子,必要時以他汀類 (statins) 降血脂。
- 第一線:血小板聚集抑制劑——阿斯匹靈 (aspirin, 100-325 mg)、氯吡格雷 (clopidogrel)、潘生丁 (dipyridamole, 3 × 400 mg);相當數量病人對阿斯匹靈與潘生丁有反應。
- 次發性 Degos 病:在原疾病標準治療外加入抗血小板藥物 (antiplatelet agents)。
- 全身性類固醇會使疾病惡化,不應處方使用;腫瘤壞死因子抑制劑英夫利昔單抗 (infliximab) 在一篇報告中無效。
- 第二線:肝素 (heparin) 及其他抗凝策略(如 warfarin、fluindione),成效不一;對抗血小板無反應的急性重症病人可試低分子量肝素 (low-molecular-weight heparin)。
- 無反應或急性重症:靜脈注射免疫球蛋白 (intravenous immunoglobulins, 1 g/kg/day 連續 2 天;或 0.4 g/kg/day 連續 5 天)。
- eculizumab(抗 C5,阻止終末補體複合體 C5b-9 生成)少數病人有顯著反應(尤其急性血栓期),但部分無反應或持續進展。
- 曲前列尼爾 (treprostinil)(前列環素類似物)在 1 名系統性硬化症/紅斑性狼瘡重疊病人及 1 名 eculizumab 治療下持續進展的 17 歲嚴重病人身上有效。
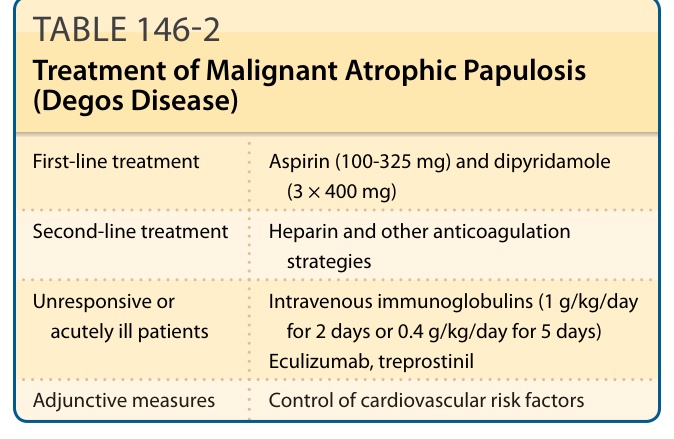
表 146-2:惡性萎縮性丘疹病的治療。